Клинические формы врожденных и наследственных заболеваний ретинального пигментного эпителия


Описание
В зависимости от топографии и локализации патологического процесса, типа наследования, возраста, в котором возникло заболевание, предложено несколько классификаций дистрофий сетчатки, затрагивающих слой РПЭ.Наблюдающиеся у детей врожденные и наследственные заболевания сетчатки, при которых отмечается патология РПЭ, включают большое количество как редких, так и более распространенных видов патологии, при которых поражаются:
- слой сосудов хороилен (хороидеремия, атрофия гирате, центральная ареолярная хороидальная дистрофия, кристалли новая ретинопатия Биетти);
- непосредственно пигментный эпителий (ретикулярная дистрофия Шегрена, макроретикулярная дистрофия Слайдера, окончатся блестящая макулярная дистрофия Доннелла, болезнь Беста, центральная ареолярная дистрофия ретинального пигментного эпителия, дистрофия Lefler — Wadsworth — Sidbury, болезнь Штаргардта, дистрофия Noble — Сагг — Siegel, врожденные альбинотические пятна, врожденные скопления пигмента в виде следа медведя);
- комплекс ретинальный пигментный эпителий — фоторецепторы (fundus flavimaculatus, белоточечный ретинит, пятнистая сетчатка и др.).Врожденные скопления пигмента в виде следа медведя представляют собой округлой или овальной формы скопления гипертрофированных клеток РПЭ с крупными гранулами пигмента. Изменения РПЭ не прогрессируют, обычно наблюдаются, окружающая сетчатка выглядит неизмененной. Наследственная природа не установлена, однако в литературе описаны семейные случаи данных изменений. Вследствие дегенерации фоторецепторов, прилегающих к гипертрофированному ПЭ, у больных выявляют дефекты в поле зрения. На флюоресцентной ангиограмме (ФАГ) видна гипофлюоресценция соответственно отложению пигмента. Отложения пигмента чаще всего видны в верхнем и нижнем темпоральном секторах. Размер пятен от 0,1 до 1,0 диаметра диска (ДД) зрительного нерва. Сосуды и диск зрительного нерва не изменены.
К «пятнистым» заболеваниям сетчатки относятся также fundus albipunc- tatus и retinitis punctata albescens. Fundus flavimaculatus будет рассмотрен в разделе «Болезнь Штаргардта».
Fundus albipunctatus — стационарное заболевание с аутосомно-рецессивным типом наследования, характерными признаками которого являются ночная слепота (никталопия) и наличие белых пятен на средней периферии глазного дна и в заднем полюсе (рис. 4.4).
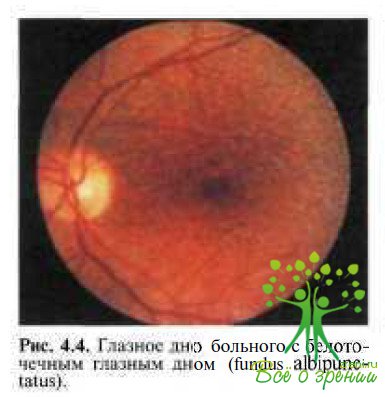
Никталопия обусловлена чрезмерно замедленным процессом регенерации зрительных пигментов. Обе фазы темновой адаптации значительно удлинены, как и при болезни Огуши. Палочковая ЭРГ значительно снижена, тогда как колбочковая обычно в пределах нормы или немного снижена. На ЭОГ практически отсутствует темновой спад, а световой подъем очень низкий после 90 мин темновой адаптации и достигает нормальных значений после пребывания в темноте в течение суток.
Retinitis punctata albescens характеризуется прогрессирующей никталопией с аутосомно-рецессивным типом наследования. На глазном дне (рис. 4.5)
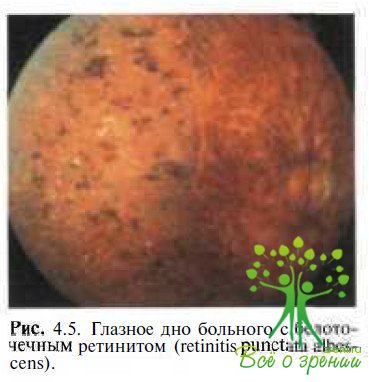
видны множественные мелкоточечные беловатые отложения. Пигментные включения на периферии указывают на генерализованный характер процесса. Темновая адаптация резко снижена, поле зрения сужено, ЭРГ или значительно снижена по амплитуде, или не регистрируется. На ЭОГ отсутствует световой подъем.
Пятнистая сетчатка Кондори — заболевание с аутосомно-рецессивным типом наследования. Для данной патологии также характерны ночная слепота, нарушение кинетики зрительных пигментов и высокая острота зрения. На глазном дне видны пятна грязно- желтого цвета с четкими границами, неправильной формы, обычно локализующиеся в области экватора. Фотопическая ЭРГ не изменена, тогда как скотопическая субнормальная, но после длительной темновой адаптации достигает нормальных значений. По клинической и функциональной симптоматике пятнистая сетчатка Кондори близка к fundus albipunctatus.
Наряду с периферическими формами поражения РПЭ выделяют формы, при которых патологический процесс локализуется в макулярной области, — дистрофии пигментного эпителия макулярной области.
Данная группа заболеваний включает ряд дистрофий, характеризующихся снижением остроты зрения, нарушением цветового зрения, изменениями фотопической ЭРГ и дегенеративными проявлениями в макулярной области. В основе их лежит патология главным образом колбочек и РПЭ.
Ретикулярная дистрофия Шегрена (Sjogren) — заболевание с аутосомно-рецессивным типом наследования, начинающееся в раннем детстве обычно асимптомно. Снижение зрения происходит поздно, темновая адаптация в течение длительного периода времени в норме. Клинически начальные изменения выглядят как темнопигментированные точки в макуле размером до 1 ДД. Ретикулярная сеть пигментированных гранул обычно выходит за пределы основного пятна, никогда не пересекая экватор. Диск зрительного нерва, сосуды сетчатки и хороидеи не изменены. В более поздних стадиях появляются друзы. Все зрительные функции сохранены. В ряде случаев отмечается снижение показателей ЭОГ. Изменения лучше всего видны на ФАГ, при этом выявляется сеть гипофлюоресценции. В межвенозных областях может наблюдаться гиперфлюореспенция. В связи с медленным прогрессированием заболевания прогноз хороший.
Микроретикулярная дистрофия Спайдера (Spider) по сравнению с дистрофией Шегрена характеризуется такими нарушениями зрительных функций, как снижение остроты зрения до 20/70, присутствие относительных центральных скотом в поле зрения, изменение цветового зрения по типу прото- или тританопии. Тип наследования ауто-сомнодоминантный, прогрессирование медленное и прогноз благоприятный. Изменений на ЭРГ, ЭОГ и при адаптометрии не обнаруживают.
На глазном дне задний полюс паутинообразно пигментирован. Изменения на ФАГ более выражены, чем при дистрофии Шегрена.
Центральная ареолярная дистрофия ретинального пигментного эпителия протекает бессимптомно, без изменений психофизиологических и электрофизиологических показателей, не прогрессирует. На глазном дне в фовеа имеются участки депигментации в виде Крапчатости. На развитых стадиях заболевания участки депигментации имеют четкие края, в центре образуются друзы. В связи с вовлечением в патологический процесс РПЭ на ФАГ соответственно участкам депигментации выявляются центральные дефекты. Тип наследования аутосомно-доминантный. Форма наследственной передачи аутосомно-доминантная, мутация гена Реripherin/RDS в локусе 6р21.2-сеn.
Практически бессимптомно протекает также окончатая блестящая макулярная дистрофия Доннелла (Боппеи), лишь в поздних стадиях появляются парацентральные скотомы, могут быть снижены амплитуды потенциалов ЭРГ и ЭОГ. Картина глазного дна достаточно характерна: желтоватый блеск с красными фенестрами в макулярной области, гипопигмептация вокруг диска зрительного нерва вследствие атрофии РПЭ. ФАГ нормальная в ранних стадиях. Тип наследования аутосомно-доминантный.
Концентрическая аннулярная (кольцевая) макулярная дистрофия в ранних стациях протекает бессимптомно или характеризуется небольшим снижением зрения и ночной слепотой. В поле зрения выявляется кольцевая парацентральная скотома. Темновая адаптация снижена, отмечается сдвиг цветового зрения в сторону дейтер- или протанопии. Изменения ЭРГ и ЭОГ нетипичны, их амплитуды могут быть как нормальными, так и сниженными. На глазном дне картина «бычьего глаза», которая обусловлена наличием очагов атрофии РПЭ круглой или овальной формы вокруг интактной сетчатки. Артерии несколько сужены, имеются небольшие парамакулярные друзы. Вследствие кольцевой атрофии РПЭ на ФАГ видно кольцо гиперфлюоресценции с центральным участком гипофлюоресценции на неизменной сетчатке и РПЭ.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Болезнь Беста (Best) (вителлиформная макулярная дистрофия), по классификации Newsome, относится к генерализованным дистрофиям РПЭ.
Дистрофия Lefler — Wadsworth — Sidbury начинается в раннем детстве, характеризуется постепенным снижением зрения до 0,1. В поле зрения появляется центральная скотома, что обусловлено друзоподобными изменениями и депигментацией РПЭ в макуле, где происходит также атрофия РПЭ и хороидальных сосудов. Полное развитие клинической картины наблюдается к 15 годам. На ФАГ видно «окно» в центральной зоне. Тип наследования аутосомно-доминантный.
Дистрофия ретикулярного пигментного эпителия Noble — Carr — Siegel развивается с рождения, тип наследования также аутосомно-доминантный. Прогноз развития болезни зависит от темпов прогрессирования патологических процессов в РПЭ. У ребенка наблюдаются нистагм и соответственно низкая острота зрения (до движения руки у лица). Данная патология у половины больных сопровождается высокой близорукостью, свыше 8,0 дптр. Фовеальный рефлекс отсутствует. В связи с атрофией РПЭ хорошо видны хороидальные сосуды, в заднем полюсе — отложения пигмента. На ФАГ определяются «окна» в области атрофированного РПЭ. В поле зрения - центральная скотома. Темповая адаптация в пределах нормы. Изменения электрофизиологических показателе й очень значительны: ЭРГ снижается до нерегистрируемой, ЭОГ уплощается.
Паттерн-дистрофия (рис. 4.6) относится к группе наследственных заболеваний РПЭ с благоприятным течением, для которых характерны гранулярные или ретикулярные пигментированные паттерны (пятна) на глазном дне. Форма наследования — аутосомно-доминантная мутация гена Peripherin/RDS в хромосомном локусе 6p21.2-cen.
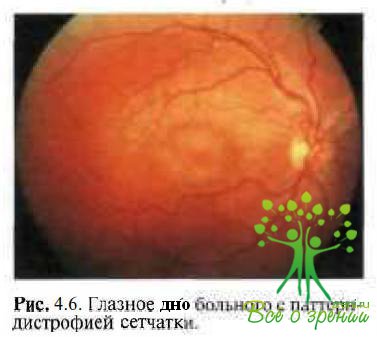
В раннем возрасте симптомы заболевания практически отсутствуют. Острота зрения может быть немного снижена вследствие изменений в фовеальной области сетчатки. Остальные зрительные функции обычно не изменены, ЭРГ также в пределах нормы. Несмотря на значительные изменения РПЭ, в большинстве случаев ЭОГ нормальная либо появляется снижение светотемнового коэффициента до 150—180 %, что никогда не наблюдается при вителлиформной дистрофии.
Болезнь Штаргардга (Stargardt) и fundus flavimaculatus. Среди многих видов наследственных макулярных дистрофий наиболее распространенными и изученными являются болезнь Штаргардта и ftindus flavimaculatus. Нередко в литературе их объединяют под термином «болезнь Штаргардта/fundus flavimaculatus», подчеркивая тем самым предполагаемое единство их происхождения и/или переход одной формы в другую по мере развития.
В начале XX в. К. Stargardt (1913) впервые описал заболевание как наследственную патологию макулярной области, развивающуюся в молодом возрасте (7—20 лет). Картина глазного дна обоих глаз характеризуется наличием о центральной зоне пигментированных круглых точек, участков депигментации и атрофией РПЭ (рис. 4.7).
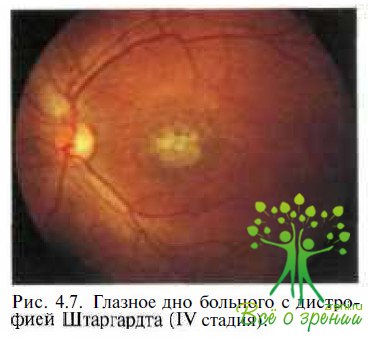
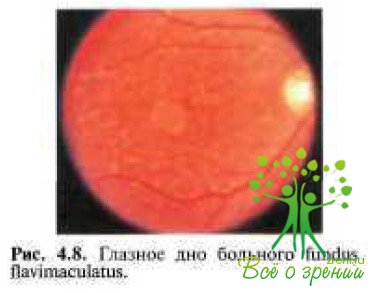
В том случае, если заболевание развивается в I—II декадах жизни, предпочитают употреблять термин «болезнь Штаргардта». Если же изменения в макулярной области и на периферии появляются в более позднем возрасте, то рекомендуют использоватъ термин «ftindus flavimaculatus» (рис. 4.8).
В зависимости от вариаций картины глазного дна предложено несколько классификаций. K.G. Noble и R.E, Сагг выделили 4 типа данного заболевания:
- — макулярная дегенерация без крапчатости;
- — макулярная дегенерация с пара- фовеальной крапчатостью;
- — макулярная дегенерация с диффузной крапчатостью;
- — диффузная крапчатостъ без макулярной дегенерации.
Острота зрения наиболее рано снижается при 1 типе, при IV же остается высокой достаточно долго. Типы II и III являются как бы комбинациями I и IV типов.
С точки зрения функциональной классификации, дистрофия Штаргардта может проявляться одним из трех выделенных синдромов; синдромом макулярной дисфункции с изолированным процессом в макулярной области; синдромом колбочковой дисфункции со снижением функции всех колбочек, как центральных, так и периферических; синдромом колбочково-палочковой дисфункции с поражением колбочковой и палочковой систем сетчатки. Оценивая различные факторы: молекулярно-генетические исследования, возраст, первые клинические проявления, длительность заболевания и клиническую картину с различной топографией процесса, показатели ЭРГ, N. Lois и соавт, (2001) выделяют три фенотипа при наследуемой по аутосомно-рецессивному типу дистрофии Штаргардта с мутацией в гене АВСЛ4.
Генетические исследования. Наиболее часто дистрофия Штаргардта наследуется по аутосомно-рецессивному типу, при котором в основном наблюдаются I и II типы изменений на глазном дне. При аутосомно-доминантном типе наследования картина глазного дна соответствует III и IV типам.
В последние годы все большее значение в диагностике наследственных заболеваний придают результатам генетических исследований. Для болезни Штаргардта определен локус вызывающего ее гена, экспрессирующегося в фоторецепторах, который был назван ABCR. Установлено, что ген ABCR идентичен по своей последовательности гену RmP, который является интегральным мембранным гликопротеином, локализующимся по краю дисков НС зрительных клеток.
Выявлено, что гены для нескольких членов суперсемейства ABCR-переносчиков вовлечены в развитие рада наследственных заболеваний сетчатки человека. Так, при аутосомно-рецессивном типе наследования болезни Штаргардта мутированный ген локализован на хромосомах 13q и р21 — р22 (STGD1), а при аутосомно-доминантной — на хромосоме 6ql4 (STGD3).
На основании результатов иммунофлюоресцентной микроскопии и вестернблоттинганализа показано, что ген ABCR присутствует в фовеальных и перифовеальных колбочках.
L.L. Molday и соавт. (2000) пришли к выводу, что мутация гена ABCR приводит к дегенерации фовеальных колбочек, вследствие чего снижается зрение при болезни Штаргардта. Более того, при обследовании семейства, члены которого были больны пигментным ретинитом и болезнью Штаргардта, G.M. Rozet и соавт. (1999) установили, что гетерозигота ость гена ABCR приводит к развитию дистрофии Штаргардта, а гомозиготность — пигментного ретинита.
Таким образом, результаты генетических исследований, проведенных в последние годы, свидетельствуют, что, несмотря на различия в клинической картине пигментного ретинита, болезни Штаргардта и fundus flavimaculatus, а также макулодистрофии, связанной с возрастом, все они являются аллельными нарушениями локуса гена ABCR
Мутации гена ABCR сопровождаются различными фснотиничсскнчи которые определенным образом зависят от специфической последовательности аминокислот. Так, при изменении последовательности в экзоне 42 гена АВСК, заключающемся в замещении глицина глутамином, отмечаются атрофические повреждения макулы, перифовеальные желтовато-белые пятна, нормальная амплитуда волн ЭРГ. Другой фенотип характеризуется темной сосудистой оболочкой, более диффузно разбросанными по глазному дну желтовато-белыми пятками, однако замены глицина на глутамин не выявлено. При выраженных атрофических изменениях РПЭ и субнормальных показателях колбочковой и палочковой ЭРГ мутации гена ABCR найдена только у 1 больного из 7.
В связи с тем, что мутации гена ABCR сопровождаются различными фенотипическими нарушениями, появляется возможность консультирования больных по поводу прогноза течения заболевания и остроты зрения, а также новые пути поиска возможных способов лечения.
Попытки корреляции генотипа и фенотипа при дистрофии Штаргардта предпринимали многие ученые, при этом учитывали локализацию мутации, ее комплексность, гомо - и гетерозиготность. Так, поданным G.Gerth и соавт. (2002), при гомозиготных мутациях в определенных парах нуклеотидов болезнь Штаргардта манифестирует в первые 5 лет и проявляется генерализованной колбочково-палочковой дисфункцией, а комплексные (на 2 участках и более) гетерозиготные мутации приводят к более позднему проявлению заболевания с менее выраженными фенотепичискеми нарушениями в виде центральной макулярной дисфункции. Однако в ряде случаев фенотипические различия не могут быть объяснены генотипически, что свидетельствует об участии в развитии болезни других, возможно, внешних факторов.
Клиническая картина. В зависимости от картины глазного дна, возраста, в котором начинается заболевание, результатов функциональных исследований S. Мегін (1993) выделив два основных типа болезни Штаргардта.
Тип I. Клинические проявления наблюдаются у детей 6—12 лет. Острота зрения прогрессивно снижается. На глазном дне в макулярной области билатерально и симметрично расположены скоплении коричневого пигмента (крапчатость), окруженные участками депигиперпигментации. Часто наблюдается вариабельность глазного дна. В ряде случаев картина напоминает «бычий глаз».
Установить правильный диагноз помогает ФАГ. Слабовыраженные изменения в макуле, выявляемые при офтальмоскопии, на ФАГ определяют по гиперфлюоресценции в месте поражения РПЭ.
По мнению ряда ученых, феномен «бычьего глаза» наблюдается почти у всех больных с болезнью Штаргардта. Вследствие скопления пигмента в макулярной области флюоресцеин невиден (картина «молчащего хороида»), вокруг темного пятна — кольцо флюоресценции, обусловленное атрофией РПЭ.
Результаты гистологического исследования свидетельствуют о значительной вариабельности размеров клеток РПЭ, чего не наблюдается и норме. Крупные клетки РПЭ образовывают гранулярную субстанцию, которая по многим свойствам соответствует видоизмененному липофусцину. Количество меланина уменьшается и он смена периферию цитоплазмы клетки в сторону наружных сегментов фоторецепторов. В более поздних стадиях заболевания часть клеток РПЭ дегенерирует. В макулярной области большая часть фоторецепторов и клеток РПЭ исчезает.
Тип II. В литературе имеются указания на более позднее начало заболевания, однако это может быть связано с более поздним обращениям пациентов к офтальмологам из-за медленного снижения остроты зрения.
Особенностью II типа является наличие множественных и распространенных желтоватых пятен на глазном дне, помимо типичных изменений в центральной области. Этим объясняются и более выраженные изменения электрофизиологических параметров. Так, в ЭРГ значительно снижены ответы палочковой системы. Ка на ЭОГ также значительно ниже, чем при I типе.
Наличие желтоватых пятен за пределами макулярной области затрудняет четкое разделение болезни Штаргардта и fundus flavimaculatus.
Как самостоятельная форма заболевания fundus flavimaculatus встречается редко, обычно желто-пятнистое глазное дно сочетается с болезнью Штаргардта.
Поскольку при типичном течении заболевай изменения на глазном дне к виде множественных сероватых или желтоватых пятен различной величины располагаются между фовеа и экватором, заболевание может протекать бессимптомно. При динамическом наблюдении за больными могут быть выявлены изменения формы, цвета, размера пятен, а также картины, выявленной на ФАГ: участки гиперфлюоресценции становятся гипофлюоресциирующими или наоборот.
Периферическое поле зрения обычно не сужено. При болезни Штаргардта выявляют относительные или абсолютные центральные скотомы, размеры которых зависят от площади поражения макулярной области. Изменениям в макулярной области сопутствуют расстройства цветового зрения по типу дейтер- или протанопии.
Вследствие изменений сетчатки за пределами макулярной области при fundus flavimaculatus отмечено повышение конечного порога снеговой чувствительности при темновой адаптометрии на несколько логарифмических единиц. У некоторых пациентов с болезнью Штаргардта отсутствует колбочковый отрезок кривой темновой адаптации.
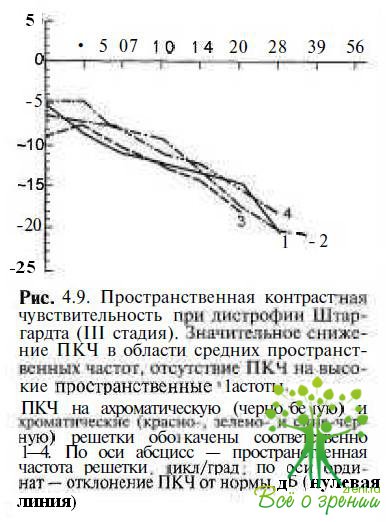
Пространственная контрастная чувствительность резко снижена в области высоких пространственных частот (рис. 4.9).
Контактная чувствительность (on – и off- активность колбочковой системы), оцениваемая по времени сенсомоторной реакции при предъявлении стимула темнее или светлее фона, отсутствует в центральной области, частично сохраняясь в зоне 10° от центра.
Наиболее информативными методами диагностики и дифференциальной диагностики заболеваний макулярной области сетчатки являются электроретинография и электроокулография. Электрофизиологические показатели (общая ЭРГ и ЭОГ) могут быть нормальными на ранних этапах развития заболевания, в то время как макулярная ЭРГ снижается и пиковая латентность b-волны удлиняется. По мере прогрессирования патологического процесса локальная ЭРГ не регистрируется (рис. 4.10),
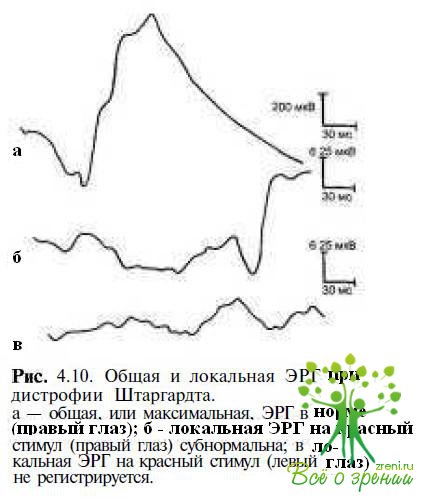
амплитуды фотопических ответов ЭРГ снижаются, показатели ЭОГ становятся умеренно субнормальными.
По мнению многих исследователей, в начальных стадиях болезни Штаргардта и fundus flavimaculatus ганцфельд-ЭРГ не изменена. Применение более тонких методических приемов электрофизиологических исследований позволило более точно выявить структуру сетчатки, изменения, обусловливающие функциональные нарушения в сетчатке.
Помимо значительного угнетения колбочковых ответов в ЭРГ, у больных с болезнью Штаргардта выявлено снижение биоэлектрических реакций наружных слоев сетчатки (фоторецепторов) в парамакулярной и периферической зонах, что свидетельствует о генерализованном поражении сетчатки обоих систем (палочковой и колбочковой) уже на ранних стадиях заболевания.
G.B.M- Moloney и соавт. (1983) у 100 % обследованных лице данной патологией выявили субнормальную колбочковую ЭРГ, а у 50 % — сниженную палочковую ЭРГ.
Прогрессирование патологического процесса сопровождается более выраженным и более частым снижением всех компонентов как локальной ЭРГ, так и ганцфелъд-ЭРГ.
Основываясь на степени угнетения различных компонентов ЭРГ, K.G. Noble и R.E. Сагг (1971), выделили 4 группы больных по стадиям заболевания. Динамическое наблюдение за больными в течение 10—14 лет позволило авторам определить наиболее неблагоприятный прогноз течения заболевания по результатам электрофизиологических исследований: снижение по сравнению с нормой фотопической Ь-волны на 43 %, скотопической на 22 %; а-волны ганцфельд-ЭРГ на 13 %, Ь-волны ганцфельд-ЭРГ на 5 % и ответов на мелькания «белого» света с частотой 32 Гц — на 21 %. ЭОГ также оказалась субнормальной, Ка составлял лишь 73,5 % от нормальных значений. Результаты электроокулографии неудивительны, так как первоначальное поражение РПЭ и фоторецепторов неизбежно должно повлечь за собой снижение светового подъема на ЭОГ.
Встречающиеся в литературе расхождения в данных о степени и частоте изменений электрофизиологических показателей, по-видимому, связаны с различиями в методических приемах, обследованием больных с разными стадиями заболевания и немногочисленностью групп пациентов.
Дифференциальная диагностика. Клиническая картина некоторых заболеваний сетчатки может быть схожа с таковой болезни Штаргардта и fundus flavimaculatus. К таким заболеваниям относятся доминантная прогрессирующая фовеальная дистрофия, колбочково-палочковая и палочко-колбочковая дистрофии, ювенильный ретиношизис, центральная ареолярная хоровдальная дистрофия, fundus albi- punctatus. Генетические и функциональные исследования помогают в дифференциальной диагностике болезни Штаргардта и fundus flavimacu- latus.
Острая задняя мультифокальная плакоидная пигментная эпителиопатия. Впервые острая задняя мультифокальная плакоидная пигментная эпителиопатия (ОЗМППЭ) была описана J.D. Gass в 1968 г. Особенностями данного заболевания являются внезапное резкое снижение остроты зрения и появление скотом в поле зрения. На глазном дне видны множественные желтовато-белые плакоидные поражения в РПЭ (рис. 4.11 ),
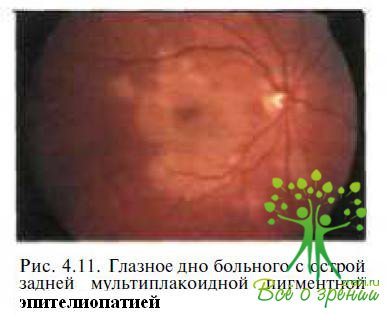
которые через несколько недель могут спонтанно исчезнуть, после чего наступает выздоровление. Вопрос о первичности поражения РПЭ или хороидальных сосудов до сих пор обсуждается в литературе. В большинстве случаев острому развитию заболеваний предшествует продромальный период (повышение температуры тела, миангия. недомогание) или наличие сопутствующей патологии (менинго-энцефалит, почечный васкулит, тиреоидит, эритема нодозум).
Результаты электрофизиологических исследований больных с ОЗМГТПЭ свидетельствуют о резко выраженных изменениях РПЭ. Ка на ЭОГ всегда низкий (110—130 % по сравнению с 185 % в норме). После выздоровления этот показатель остается ниже нормы. В остром периоде скотопическая ЭРГ также в несколько раз ниже нормальной и нормализуется после реконвалесценции.
Эти данные, особенно результаты окулографии, свидетельствуют о первичном поражении ПЭ. Согласно результатам флюоресцентной ангиографии, по мнению ряда исследователей, первоначально патологический процесс возникает в хороидее. В связи с тем, что ФАГ имеет ряд ограничений, связанных с невозможностью оценить состояние хориокапилляров и более крупных хороидальных сосудов из-за маскировки их слоем РПЭ и ксантофиллами макулярной области, в подобных случаях наиболее информативным методом является ангиография с индоцианином зеленым (ICG).
При ICG пик возбуждения (805 нм) и пик флюоресценции (835 нм) находятся около инфракрасной области спектра, для которой почти «прозрачны» РПЭ, меланин, макулярный ксантофилл и хороидальный пигмент. Более того, поскольку индоцианин зеленый полностью связан с белком, он не выходит через фенестры хориокапилляров.
При ОЗМППЭ с помощью ICG выявляют значительную задержку заполнения хороидеи красителем, а также обширные области неперфузируемых хоровдальных артериол. Распространенность неперфузируемых хороидальных сосудов позволяет предположить, что в патологический процесс вовлечены более крупные задние ресничные артерии. По мнению исследователей, эти данные свидетельствуют о первичности нарушений перфузии хороидальных сосудов при ОЗМППЭ и вторичное вовлечение в патологический процесс РПЭ.
ОЗМППЭ в большом числе случаев сочетается с васкулитами ряда органов. Предполагают, что именно воспаление сосудов глаза клинически проявляется как ОЗМППЭ.
Хороидеремия. Установлено, что хороидеремией болеют только мужчины, а передают заболевание только женщины. У женщин — носительниц патологического гена, как правило, не наблюдается снижения зрения, а изменения на глазном дне обычно слабо выражены или отсутствуют. Согласно закону Менделя, у половины сыновей женщин — носительниц гена хорандеремии наблюдается развитая картина заболевания.
Генетические аспекты. Хороидеремия — заболевание, наследуемое по Х-сцепленному с полом типу, при котором женщины — носительницы патологического гена передают его сыновьям. Аллель связан с Х-хромосомой. На основании результатов обследования членов 3 семей с хороидеремией R.A. Lewis и соавт. (1985) предположили, что локус хороидеремии может быть фланкирован маркерами DXYSI и DXSI1, ближе к DXYS1, на Х-хромосоме. В генетических исследованиях установлено, что наиболее возможным местоположением гена хороидеремии является локус на Xq21.2.
Клинические проявления. Хороидеремия характеризуется двусторонней диффузной прогрессирующей дегенерацией РПЭ и хориокапилляров. Средний возраст пациентов с видимыми проявлениями заболевания 31 год, при этом еще задолго до обращения к специалисту (в течение 20 лет) больные отмечают симптомы ночной слепоты и сужение полей зрения.
У больных с хороидеремией уже в детском возрасте на глазном дне во всех квадрантах отмечаются рассеянные пигментные пятна с островками атрофии на средней периферии, через которые может быть видна склера, истончение хороидеи, смешение пигмента в слои сетчатки ПО ходу сосудов (рис. 4.12).
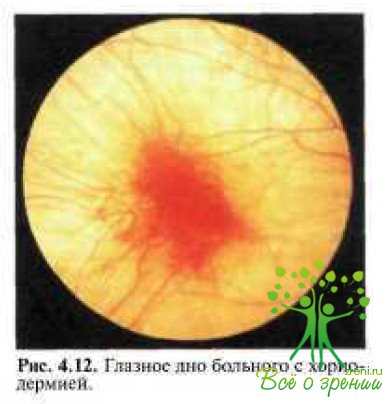
В отличие от пигментного ретинита при хороидеремии сужение ретинальных артерий незначительное, вены обычно не изменены. Побледнение диска зрительного нерва и темпоральная атрофия наблюдаются у 29 % больных. Заболевание может возникнуть в раннем детском возрасте. В дальнейшем изменения на глазном дне прогрессируют и могут привести к слепоте уже к 30 годам, если патология хороидеи и РПЭ рано распространяется на макулярную область сетчатки.
У некоторых больных хороидеремия может сочетаться с деформациями скелета, умственной отсталостью, колобомой радужки.
Зрительные функции. Острота зрения у больных с хороидеремией обычно долго сохраняется высокой, несмотря на прогрессирующее сужение полей зрения. В процессе развития заболевания в поле зрения выявляют множественные скотомы на участках 25—50° от точки фиксации соответственно зонам поражения на глазном дне. Слепое пятно расширено. В дальнейшем образуется кольцевая скотома. В то же время происходит постепенное сужение поля зрения до точки фиксации 3° или меньше.
Цветовое зрение может оставаться нормальным или наблюдается тританопия — тританомалия.
У больных с раннего детства ухудшаются показатели темновой адаптации. В начале заболевания скотопическая часть кривой темновой адаптации снижается на 1—2 лог. ед., в дальнейшем она может отсутствовать. У больных пожилого возраста снижается и фотопическая чувствительность.
Флюоресцентная ангиография. Картина на ФАГ при хороидеремии достаточно типична. Собственные сосуды сетчатки обычно не изменены. В то же время вследствие дегенерации РПЭ видно просачивание флюоресцеина из хороидеи и усиление гиперфлюоресценции вокруг ее непораженных участков. В зонах, в которых отсутствуют хориокапилляры, наблюдается гипофлюоресценция. Продолжительность хороидеальной и ретинальной фаз увеличена так же, как и интервал между фазами наполнения ретинальных артерий и вен.
Электроретиногрифия. Для хороидеремии характерно значительное угнетение волн ЭРГ, прежде всего ее скотопических компонентов. У взрослых больных скотопическая ЭРГ либо не регистрируется, либо имеет характер микроЭРГ. В то же время отмечается удлинение периода развития фотопических компонентов ЭРГ при сохранении их амплитудных значений в пределах нормы.
При световой микроскопии в зонах гипопигментации виден тонкий слой атрофированной сетчатки, клеточными элементами которого, по-видимому, являются биполярные и ганглиозные клетки, а также нервные волокна. На средней периферии не обнаруживают фоторецепторов. В области макулы и около диска зрительного нерва все слои сетчатки, кроме фоторецепторов, остаются более сохранными. В зоне гипопигментации РПЭ и хоровдея отсутствуют. В то же время в макулярной области РПЭ сетчатки практически не изменен. Хориокапилляры и хороидея сохранены на тех же участках, где и РПЭ сетчатки.
Более детально изучены структурные изменения тканей глаза при хороидеремии с помощью электронной микроскопии, на участках сетчатки, на которых сохранены все клеточные элементы, отмечается дезорганизация слоя палочек и колбочек, причем первые изменены более значительно, поскольку их наружные сегменты в большом количестве поглощаются макрофагами. РПЭ выявляется только на участках, где сохранены сетчатка, мембрана Бруха и хориокапилляры.
Нарушения в слое фоторецепторов и РПЭ не сопровождаются изменением фоторецепторной активности и концентрации цГМФ. До сих пор остается дискутабельным вопрос о первичности поражения структур при хороидсремии. Атрофия хороидеи и пигментного эпителия — наиболее выраженные клинические проявления этого наследственного заболевания. Предполагают, что механизмы развития хороидеремии и пигментного ретинита могут быть сходными.
Биохимические исследования, проведенные с целью определения содержания интерфоторецепторного ретинолсвязывающего белка, циклических нуклеотидов (цАМФ, цГМФ), уровня фагоцитарной активности при хороидеремии и пигментном ретините, позволили определить новые стороны патогенеза данных заболеваний, однако непосредственно установить причины развития пигментного ретинита и, тем более, хороидеремии не удалось.
Большой интерес представляют работы, в которых представлены результаты обследования носителей гена хороидеремии, имеющие значение как для диагностики заболевания у лиц мужского пола, так и для прогнозирования возможности передачи хороидеремии последующим поколениям.
Офтальмоскопическая картина, характерная для хороидеремии, у женщин, носительниц патологического гена, выражена в значительно меньшей степени, чем у их больных сыновей.
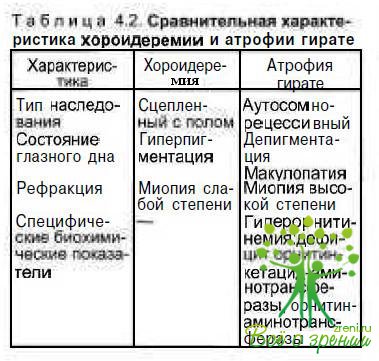
Дифференциальная диагностика. По клинической картине с хороидеремией наиболее сходна атрофия гирате. Различия заключаются в типе наследования: хороидеремия сцеплена с полом, атрофия гирате наследуется по сомно-рецессивному типу (табл. 4.2).
Методы лечения хороидеремии не разработаны.
Атрофия гирате. Среди заболеваний, характеризующихся генерализованной атрофией хороидеи и сетчатки, атрофия гирате выделяется своими особенностями: фестончатыми изменениями на глазном дне, гиперорнитинемией, дефицитом фермента — орнитинами нотрансферазы. Тип наследования аутосомно-рецессивный. Заболевание проявляется в первой декаде жизни в виде округлых очагов полной атрофии хороидеи и сетчатки по средней и дальней периферии. С возрастом эти очаги увеличиваются, сливаются и приближаются к интактной области заднего полюса глаза. Наиболее ранние проявления атрофии гирате — отсутствие периферического зрения и ночная слепота. Распространение атрофических очагов к макулярной области приводит ЭОГ на ранних этапах развития заболевания остается нормальной, тогда как ЭРГ практически исчезает (рис. 4.13).
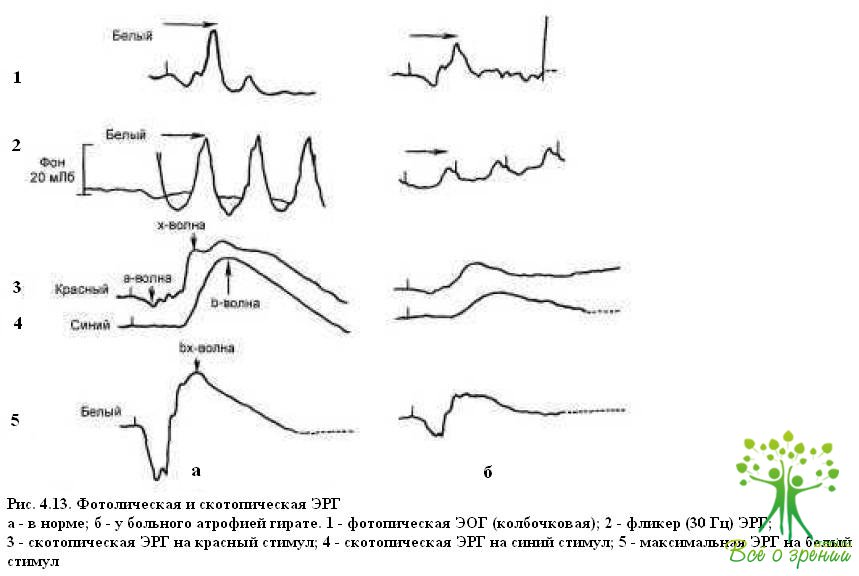
Характерным симптомом атрофии гирате является изменение кривой тем- новой адаптации (рис. 4.14).

Патогенез. Первыми об увеличении содержания орнитина в плазме крови х больных с атрофией гирате сообщили O.Simell и K.Takki (1973). Его уровень превышал нормальные значения в 10—20 раз. Позднее высокий уровень орнитина был обнаружен у больных и в других жидкостях организма: цереброспинальной и внутриглазной .
Ряд исследователей установили, что у части больных с атрофией гирате при лечении витамином В6 (пиридоксин) наблюдается снижение уровня орнитина в плазме, тогда как у других пациентов с аналогичной картиной заболевания этого не происходит. Это может быть связано с наличием двух генетических типов данного заболевания.
К настоящему времени выделены три типа атрофии гирате: чувствительная и нечувствительная к пиридокси- ну, а также с нормальным уровнем орнитина.
Повышенный уровень орнитина при атрофии гирате объясняют дефицитом орнитинаминотрансферазы в фибробластах и лимфоцитах. Данный фермент необхо¬дим для катаболизма орнитина в глутамат и пролин, которые затем присоединяются к протеину. Более того, у больных с атрофией гирате уровень глутамата, глутамина, креатина и лизина в плазме крайне низкий. Считают, что описанные выше биохимические сдвиги являются причиной развития мышечной дистрофии у больных с атрофией гирате. Низкий уровень орнитинаминотрансферазы обнаружен также в эпителиальных клетках ресничного тела, радужке, хрусталике и пигментном эпителии что объясняет первичное поражение пигментного эпителия сетчатки и следующую за ним вторичную атрофию хороидеи.
Эта точка зрения была подтверждена результатами экспериментальных исследований. Несмотря на то что до настоящего времени неизвестен патогенетический механизм развития атрофии гирате вследствие дефицита орнитинаминотрансферазы, предполагают, что первичное поражение развивается именно в пигментном эпителии сетчатки. Данное предположение подтверждают результаты исследования у больных с атрофией гирате с-волны ЭРГ, которая, как известно, отражает состояние именно пигментного эпителия. Эту волну регистрируют только у больных с самыми начальными проявлениями заболевания, в развитых стадиях эта волна отсутствует.
Генетические аспекты. Атрофия гирате имеет аутосомно-рецессивный тип наследования с двумя различными генетическими формами: чувствительной и нечувствительной к витамину. Работами цито- и молекулярных генетиков был установлен локус атрофии гирате на хромосоме 10. Позднее 2 локуса, ответственных за ген орнитинаминотрансферазы, были обнаружены у дистального конца длинного плеча 10 хромосомы.
Предполагают, что обе формы атрофии гирате являются результатом мутаций орнитинаминотрансферазы.
Лечение. Знание биохимических нарушений при атрофии гирате позволило предпринять попытки снизить уровень орнитина в крови с помощью диеты с низким содержанием аргинина и протеина.
Применение лизина, креатина и другие способы нормализации биохимических показателей кропи не оказывали влияния на развитие и прогрессирование глазных проявлений у больных с атрофией гирате.
Хотя лечение заболевания невозможно, имеются способы генетического прогнозирования рождения ребенка с патологией. Для сибсов больных риск заболеть составляет 2,5 %. Гетерозигот можно выявить при тестировании активности орнитинам и нотрансферазы. Возможна также пренатальная диагностика атрофии гирате по клеткам амниотической жидкости.
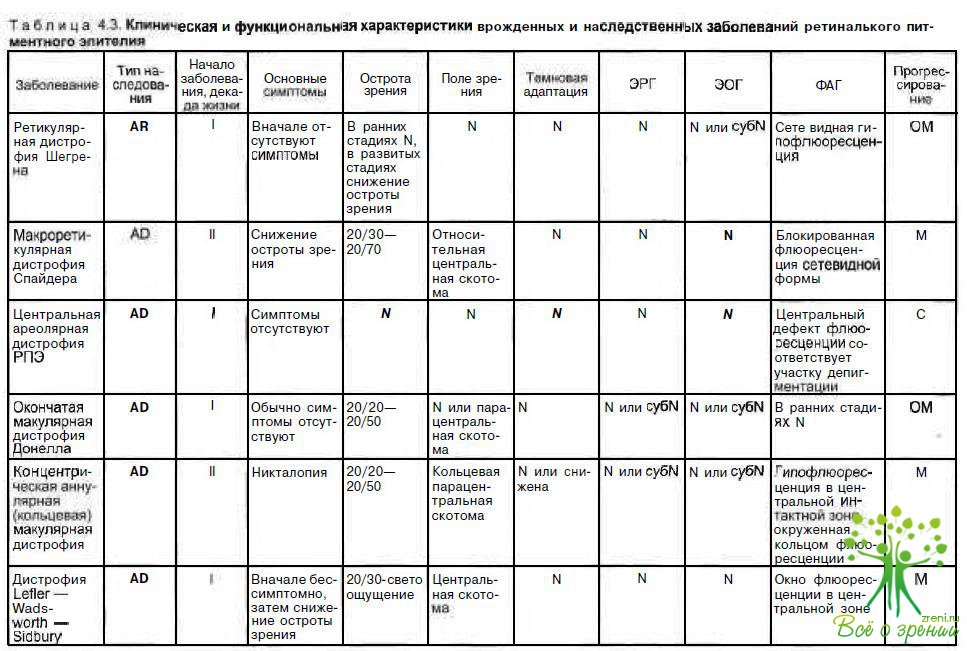
В табл. 4.3 суммированы основные признаки заболеваний, описанных в данной главе.
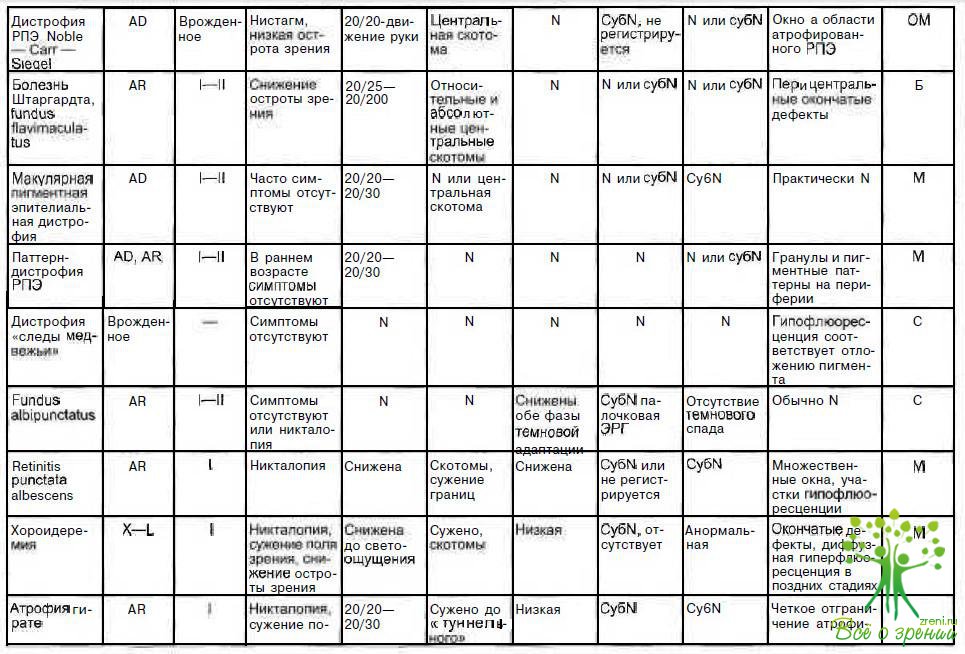
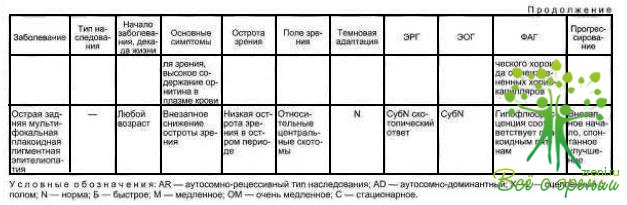
Таким образом, клиническая картина, результаты функциональных исследований, типы наследования при заболеваниях РПЭ различные. При всех описанных видах патологии РПЭ, за исключением атрофии гирате, лечения не существует. Снижение тем или иным способом содержания орнитина плазмы у больных атрофией гирате не приводит к выздоровлению, а лишь нивелирует одно из проявлений патологии.
Только новые достижения генетиков могут помочь больным с наследственными и врожденными заболеваниями, в том числе глаз. Прогресс в этой области науки вселяет надежду на то, что будут найдены подходы к патогенетическому лечению наследственных заболеваний и коррекции мутированных генов.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0