Митохондриальная патология органа зрения у детей


Описание
К митохондриальным заболеваниям относят группу гетерогенных патологических состояний, обусловленных дефектами структуры и функции митохондрий.Такие митохондриальные заболевания, как прогрессирующая офтальмоплегия и прогрессирующая офтальмоплегия с миопатией, были известны с 1962 г.
Этиология и генетические исследования. Особенно важным в понимании этиологии и патогенеза этой патологии является открытие в 1963 г. митохондриальной ДНК (мДНК), которая имеет двухнитчатую структуру. Нуклеотидная последовательность мДНК состоит из 16 569 пар нуклеотидов, на которых расположено 37 генов (рис.12.1).
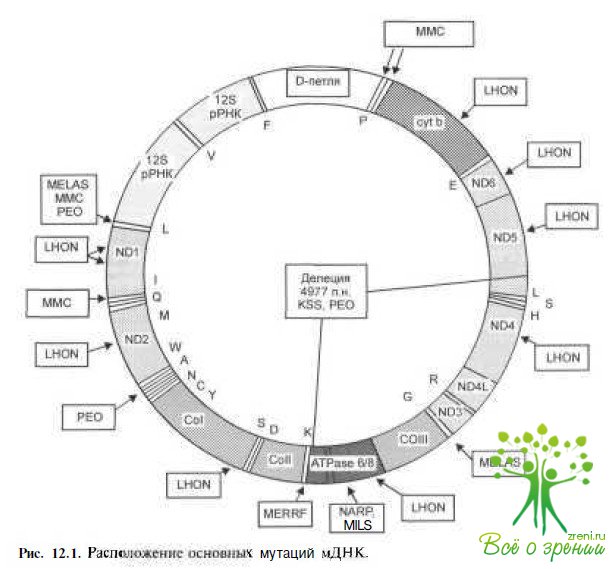
Каждая клетка включает от нескольких сотен до 10 000 митохондрий. Одна митохондрия содержит от 2 до 10 молекул ДНК. Комплексы дыхательной цепи митохондрий (КДЦМ) находятся пол двойным генетическим контролем; 13 полипептидов КДЦМ кодируются МДНК и около 80 полипептидов КДЦМ — ядерной ДНК.
Генные мутации вызывают нарушение функции КДЦМ, что приводит к нарушению синтеза АТФ и увеличению уровня свободных радикалов в клетке (агрессивных форм кислорода). За этим следуют нарушения основных жизненно важных процессов клеточного обмена — высвобождение энергии органических веществ и аккумуляция се в виде макроэргических фосфатных соединений, происходящих в митохондриях (метаболизм пирувата ?-окисление, превращения в цикле Кребса, транспорт электронов в дыхательной цепи)
Мутантная и нормальная ДНК митохондрий могут существовать в одной и той же клетке. Это состояние называется гетероплазмией. Доля мутантной мДНК, влекущей за собой значительное снижение энергетического метаболизма клетки какого-либо органа, вызывающее патологические признаки в данном органе, служит порогом фенотипической экспрессии и явдиагностическим признаком и заболевания.
Системные проявления. Для этой группы заболеваний характерны выраженный внутрисемейный, Внутри популяционный клинический полиморфизм и сочетание поражений нескольких органов и систем, которые зависят от случайного тканевого распределения мутанта их митохондрий и степени гетероплазмии в различных клетках и тканях. Так, например, при превышении порогового уровня мутации А3423С у больных может появиться один из синдромов: MELAS, KSS, MERRF. Сложность, МНОГОКОМПОНЕНТНОСТЬ биоэнергетических процессов и двойственность кодирования (ядерной и митохондриальной ДНК) отдельных этапов тканевого дыхания объясняют неспецифичность отдельных клинических проявлений, а причинами сходных клинических признаков могут быть различные нарушения энергетического обмена, обусловленные различными мутациями.
Важной особенностью митохондриляется цитоплазматичный тип наследования. Заболевание наследуется по материнской линии, оба пола болеют одинаково часто. Патологические состояния энергетического обмена предаются по аутосомно-рецессивному, аутосомно-доминантному и Х-сцепленно-рецессивному типам наследования. Десять лет назад был определен новый класс митохондриальных заболеваний – появление множественных делеций мДНК (уменьшение или отсутствие копий) как результат мутаций в ядерном геноме.
Наиболее чувствительны к нарушению активности энергетического обмена клеток центральная нервная система, орган зрения, миокард, скелетные мышцы, эндокринная система, почки, печень.
Офтальмологические проявления. Страдают в первую очередь зрительный нерв, сетчатка, глазодвигательные нервы, мышцы придаточного аппарата глаза как наиболее энергоемкие ткани. P.R. Smith и соавт. установили, что у 77 % больных с митохондриальной патологией наблюдается пигментный ретинит, у 23 % — гемеролопия, снижение зрения, фотофобия. В 44 % случаев изменялись показатели электроокулограммы, свидетельствующие о дистрофии пигментного эпителия сетчатки.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Клинические признаки митохондриальных заболеваний могут проявлятель изолированными нарушениями какого-либо отдельного органа — зрительного нерва при нейропатии Лебера аппарата при прогрессирующей наружной офтальмоплегии.
Однако для заболеваний этой группы, манифестирующих в детском возрасте, более характерны нарушения органа зрения в сочетании с патологией нескольких органов и систем, причем большое разнообразие клинических форм.
Типичная клиническая картина наследственной оптической нейропатии Лебера возникает при пороге фенотииической экспрессии мутантной мДНК 80-90 %
Неврологические нарушения отмечены в 20— 59 % случаев, у ряда больных определяют дизартрию, Патологические рефлексы, спастичностъ, слабо выраженную мозжечковую атаксию, нарушения сердечной проводимости.
Описаны мутации в генах ПД4, ПЛ. ЦД6, приводящие к развитию у этих больных мышечной дистонии или рассеянного склероза. Сроки манифестации и клинический полиморфизм зависят от особенностей мутации мДНК и выраженности гетероплазмии.
Хроническая прогрессирующая наружная офтальмоплегия у детей обычно является проявлением одного из синдромов, но не самостоятельной нозологической единицей.
Одиночные делеции мДНК выявляют у 40—50 % больных. Известны случаи этого заболевания, вызванные множественными делениями мДНК, которые контролируются ядерными генами, локализующимися на хромосомах 15q25, 10q24, 4q35. Тип наследования патологии в этих семьях аутосомно-доминантный и аутосомно-рецессивный, сроки манифестации патологического признака варьируют от 14 до 67 лет. В этих случаях отмечают сочетание двусторонней наружной офтальмоплегии с поздним появлением мышечной слабости или другими соматическими нарушениями.
В литературе нет данных о частоте и клинических особенностях изолированной прогрессирующей наружной офтальмоплегии. Эта патология у детей появляется в раннем детстве, начиная с 9 мес. Обычно первым признаком заболевания является билатеральный, симметричный, прогрессирующий ПТОЗ верхних век, признаки ремиссии могут отсутствовать. Описаны случаи одностороннего и асимметричного поражения век. Со временем присоединяются нарушения глазодвигательных мышц, возникает паретическое косоглазие.
Наружная офтальмоплегия является одним из ведущих признаков синдромов Кернса—Сейера, MELAS, ARCO, митохондриальной энцефалопатии, болезни Лея.
Пигментная дистрофия сетчатки описана при синдромах Кернса—Сейера, MELAS, NARP, болезни Лея. Клинический тип абиотрофии сетчатки чаще всего не указывается. Имеются единичные описания клинических особенностей изменений сетчатки и хороидеи.
Атрофия зрительных нервов встречается при синдромах Кернса—Сейера, MELAS, DIDMOAD, MERRF, болезни Лея
Помутнения хрусталиков отмечены при синдромах MELAS, Кернса—Сейера.
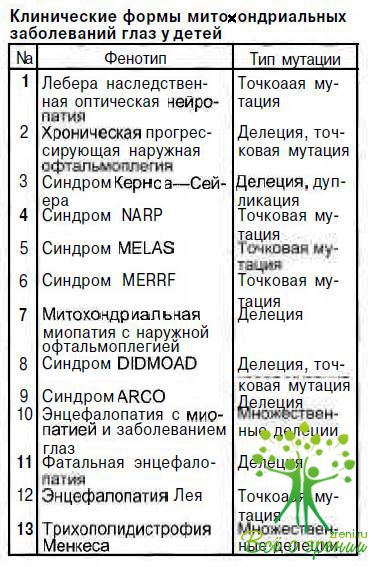
Представляем наиболее часто встречающиеся в детском возрасте митохондриальные синдромы с патологией органа зрения.
Синдром Кернса—Сейера (KSS) впервые описан T.P.Kearns и G.P.Sayre в 1958 г. К настоящему времени опубликованы сведения о нескольких сотнях больных. Подавляющее большинство случаев спорадические, однако имеется и семейное накопление заболевания. У 80—90 % пациентов определены единичные обширные делеции мДНК, локализация их может быть различной. Известны случаи сочетания делеции с увеличением или уменьшением количества копий мДНК.
Пороговые значения гетероплазмии составляют 60-80 %.
Сроки манифестации патологических признаков варьируют от 4 до 18 лет. Девочки и мальчики болеют одинаково часто. Дети при рождении нормальны. Заболевание характеризуется птозом верхних век, прогрессирующей наружной офтальмоплегией, задержкой роста. Затем появляются пигментная дистрофия сетчатки, атриовентрикулярная блокада, мышечная гипотония, мозжечковая атаксия, гилогонадизм, иногда снижение аппетита.
Птоз верхних век — наиболее типичная патология синдрома — обычно двусторонний и симметричный, параллельно ему появляются признаки медленно прогрессирующей наружной офтальмоплегии, иногда возможна диплопия. В большинстве случаев на глазном дне определяют дистрофию заднего полюса сетчатки по типу «соль с перцем» без «костных телец», могут быть дефекты пигментного эпителия сетчатки. Наблюдаются также бледный диск зрительного нерва и сужение ретинальных сосудов. В отдельных случаях заболевание протекает без дистрофии сетчатки, но с поражением хориокапиллярного слоя. Острота зрения постепенно уменьшается, появляются дефекты полей зрения, снижение амплитудных показателей электроретинограммы, прогрессирует помутнение хрусталиков.
Мышечная слабость становится ощутимой больными через несколько лет после появления птоза верхних век. Наиболее типичен нисходящий характер миопатии. Обращают на себя внимание изменение тембра голоса у детей, маскообразное, гипомимичное лицо. Половину пациентов характеризует умственная отсталость. Встречаются также полинейропатия, нейросенсорная тугоухость.
A.Engeiи соавт. описали полный и неполный синдромы Кернса—Сейера. Полный включает прогрессирующий птоз верхних век, наружную офтальмоплегию, пигментную дистрофию сетчатки и атриовентрикулярную блокаду. Частыми признаками заболевания являются поражения нервной и эндокринной систем. Начинается заболевание до 10 лет и имеет неблагоприятный прогноз.
Авторы выделяют два варианта неполного синдрома:
- сочетание хронической наружной прогрессирующей офтальмоплегии и миопатического комплекса, имеющего тенденцию к генерализации по нисходящему типу. Кроме того, может наблюдаться один из необязательных признаков;
- наличие только хронической прогрессирующей наружной офтальмоплегии. Этот вариант возникает в более старшем возрасте и встречается крайне редко.
Большое значение для диагностики имеют описания атипичных вариантов заболевания. Описана семья с манифестацией патологии у мальчика 3 лет по типу синдрома Лоу: отставание в психомоторном развитии, почечные изменения и катаракта. Клиническая картина, характерная для синдрома Кернса—Сейера, начала развиваться у ребенка к 11 годам.
Синдром MELAS — симптомокомплекс, включающий митохондриальную энцефалопатию, лактатацидоз, инсультоподобные эпизоды, впервые выделен в самостоятельное заболевание в 1984 г. Описано около 120 больных. Установлены точковые мутации мДНК в 80—90 % случаев в нуклеотоде 3243. Кроме того, описаны мутации в нуклеотвдах 3271 и 1084. Наследуется патология по цитоплазматическому типу. Пороговый эффект экспрессии признаков заболевания составляет 93— 96 %. При гетероплазмии у 62—84 % наблюдаются стертые формы заболевания.
Возраст начала, развития клинических признаков заболевания варьирует от 6 до 10 лет. Описаны случаи манифестации этого синдрома до 2 лет и в 21-40 лет. До дебюта заболевания в 90—100 % случаев дети растут и развиваются нормально.
Синдром выражается появлением судорог, рецидивирующих ГОЛОВНЫХ болей, рвоты, анорексии. Основные проявления патологии включают непереносимость физических нагрузок, инсультоподобные эпизоды, миоклонусы, глухоту. Деменция наблюдается в 90 % случаев, мозжечковые симптомы — в 33 %, атрофия зрительных нервов — в 20 %, пигментная дистрофия сетчатки — в 16 % и прогрессирующая офтальмоплегия — в 13 % случаев.
S.M. Sue и соавт. описали в 14 семьях с мутацией в нуклеотиде 3243, вызвавшей синдром MELAS, пигментные изменения сетчатки — симметричные, чередующиеся с участками депигментации, расположенные в заднем полюсе глаз и на средней периферии, а у 10 больных — атрофию зрительных нервов.
Y. Isashiki описал юношескую катаракту у одного больного с синдромом MELAS.
Некоторые пациенты имеют гемианопсию. Течение заболевания прогрессирующее.
Синдром MERRF —миоклонус-эпилепсия с «рваными» красными волокнами — впервые описан в 1980 г. N.Fukuhara. В литературе имеются сведения примерно о 50 случаях заболевания, обусловленного точковой мутацией мДНК чаще в нуклеотиде 8344, в единичных случаях — в нуклеотиде 8356. Наследуется по цитоплазматическому типу.
Следует отметить значительный внутрисемейный клинический полиморфизм, наличие полных и стертых форм заболевания, обусловленных степенью гетероплазмии. Возраст манифестации патологических признаков чрезвычайно вариабелен — от 3 до 65 лет. Ранние признаки заболевания характерны для энцефаломиопатии (утомление при физических нагрузках, снижение памяти). Типичная клиническая картина включает прогрессирующую миоклонус-эпилепсию, атаксию, деменцию, нейросенсорную глухоту, низкий рост, в 29 % случаев наблюдается атрофия зрительных нервов. Y,Isashiki обнаружил пигментную дистрофию сетчатки у трех больных.
Течение заболевания прогрессирующее.
Синдром NARP, включающей нейтропатию, атаксию, пигментный ретинит, впервые описан в 1990 г. В настоящее время известно примерно 10 больных. Патологический процесс развивается в результате точечной мутации в нуклеотиде 8993 мДНК, передается по цитоплазматическому типу. Этиопатогенез этого заболевания слабо изучен. Сроки манифестации значительно вариабельны.
Заболевание характеризуется нейрогенной мышечной слабостью, атаксией и пигментной дистрофией сетчатки. Кроме того, могут быть судороги, задержка психомоторного развития, спастические изменения мышц конечностей.
C.Y. Tsao и соавт. отмечают, что у 8 членов семьи с высоким уровнем гетероплазмии, мутацией мДНК в нуклеотиде 8993 не было типичных признаков синдрома PARP и пигментной дистрофии сетчатки.
P.F. Chinnery и соавт. описали семью с выраженным клиническим полиморфизмом этого заболевания. У матери не отмечено болезни глаз, у дочери обнаружена типичная колбочковая дистрофия, а у сына — колбочко-палочковая дистрофия сетчатки.
Течение патологического процесса прогрессирующее.
Подострая некротизирующая энцефаммттапин: Лея впервые описана D. Leigh в 1951 г. В настоящее время известно более 100 больных. Это гетерогенное патологическое состояние, обусловленное точковыми мутациями мДНК в нуклеотиде 8993, а также мутациями ядерной ДНК, приводящими к изменениям функции митохондрий.
У некоторых больных установлена мутация в нуклеотиде 8344, характерная для синдрома MERRF. Порог фенотипической экспрессии составляет 70—95 %.
Авторы высказывают предположение, что болезнь Лея — одна из наиболее тяжелых форм митохондриальной энцефаломиопатии — развивается в случае появления в клетках подавляющего большинства мутантной мДНК. Заболевание наследуется цитоплазматически, по аутосомно-рецессивному и Х-сцепленно-рецессивному типам.
Первые признаки заболевания могут быть обнаружены на 1—3-м годах жизни, известны и более поздние формы манифестации — с 7 лет. У ребенка появляется задержка психомоторного развития, рвота, снижение аппетита, различные нарушения мышечного тонуса, затем начинаются тоникоклонические и миоклонические судороги, координаторные расстройства, сонливость, снижение сухожильных рефлексов, нарастают симптомы пирамидной и экстрапирамидной недостаточности, нарушение глотания. У многих детей отмечают птоз верхних век, наружную офтальмоплегию, атрофию зрительных нервов, иногда пигментную дистрофию сетчатки.
Течение заболевания может быть хроническим, подострым и остроразвиваюшимся с быстрым фатальным течением.
Трихополидистрофия Менкеса — болезнь «курчавых волос», описана в 1962 г. I.N. Menkes, На сегодняшний день известно около 200 больных. Патологический процесс развивается вследствие мутации гена, расположенного на хромосоме Xql3.3, контролирующего функцию митохондрий по транспорту меди из клетки во внеклеточное пространство.
Клинические признаки заболевания проявляются с первых недель жизни мальчиков гипотермией, недостаточной прибавкой массы тела. Затем начинаются генерализованные судороги, миоклонические подергивания лица или конечностей, мышечная гипотония сменяется спастическими парезами. Волосы у пациентов светлые, ломкие, курчавые, редкие, кожа сухая, бледная; могут быть переломы трубчатых костей.
На глазном дне у таких больных определяют атрофию зрительных нервов, микрокисты сетчатки.
Течение заболевания прогрессирующее, приводит к фатальному исходу на 1—3-М годах жизни ребенка от субдурального кровоизлияния или септического состояния. Редко наблюдаются атипичные формы болезни Менкеса с поздним началом и продолжительностью жизни до 13 лет.
Синдром DIDMOAD - симптомокомплекс, включающий диабет несахарный, диабет сахарный, оптическую атрофию, глухоту. Заболевание характеризуется прогрессирующим развитием всех перечисленных симптомов; гетерогенное, изучено плохо. Возникает при нарушении процессов репликации и транскрипции мДНК, находящихся под контролем ядерной ДНК, наследуется по аутосомно-рецессивному типу.
Синдром АRСО — аутосомно-рецессивная кардиомиопатия с наружной офтальмоплегией. Заболевание обусловлено мутациями ядерного генома, приводящими к делеции мДНК. Клиникомолекулярные особенности патологии недостаточно изучены.
Таким образом, в литературе накоплен относительно небольшой опыт по изучению этиологии, патогенеза и клинических особенностей митохондриальных заболеваний глаз у детей. Это связано с тем, что митохондриальная патология органа зрения, манифестирующая до 15 лет, как правило, входит в группу гетерогенных заболеваний с разными типами наследования, вовлечением в патологический процесс нескольких органов и систем.
Вместе с тем нередко изменения органа зрения предшествуют появлению тяжелой клинической картины инвалидизирующих заболеваний. Это повышает роль ранней и дифференциальной диагностики митохондриальной патологии для направления на обследование в специализированные педиатрические центры.
Митохондриальную патологию следует дифференцировать прежде всего от заболеваний, вызванных аномалиями обмена органических кислот, которые наследуются по аутосомно-рецессивному типу и обусловлены мутациями ядерных ДНК.
Клинические картины заболеваний, вызванных мутациями мДНК и органическими ацидемиями, очень схожи, однако органические ацидемии манифестируют в более раннем возрасте, птоз верхних век наблюдается только у 8 % больных и заметно усиливается после физической нагрузки, между приемами пищи; наружной офтальмоплегии, как правило, не бывает.
В основном дифференциальную диагностику возможно провести на биохимическом и морфологическом уровнях.
Ранняя этиопатогенетическая диагностика митохондриальной патологии органа зрения необходима для назначения лечения, медико-генетического консультирования семьи, а в некоторых случаях и организации пренатальной диагностики.
Для лечения митохоцдриальной патологии у детей используют следующие препараты:
- «Коэнзим 0» («убихинон») — естественный переносчик электронов от 1, 2 и 3-го комплексов дыхательной цепи; дополнительное введение его повышает активность дыхательной цепи митохондрий;
- препараты янтарной кислоты («Янтавит», «Лимонтар») транспортируют электроны ко 2-му комплексу дыхательной цепи, активизируют окислительные функции митохондрий, оказывают выраженный антиоксидантный эффект;
- «Цетохром С» — одна из ключевых субстанций дыхательной цепи митохондрий, принимает участие в переносе электронов от 3-го к 4-му комплексу;
- «Никотинамид» выполняет функцию промежуточного переносчика восстановительных эквивалентов от различных субстратов в окислительно-восстановительных процессах;
- «Рибофлавин» — активный структурный элемент более 30 респираторных флавопротевдов. Фдавиновые ферменты играют роль первичных детодрогеназ при окислении сукцината и жирных кислот;
- «Тиамин», или липоевая кислота, является составной частью ко ферментов (Е1 и Е2), которые участвуют в обмене углеводов, аминокислот и функции цикла Кребса;
- «Биотин» — кофермент ряда кокарбоксилаз, участвующих в метаболизме пировиноградной кислоты;
- «Карнитин», его главная функция — перенос жирных кислот через митохондриальную мембрану — влияет на энергетический метаболизм внутри митохондрий;
- «Димефосфон» активизирует тканевое дыхание и стабилизирует состояние клеточных мембран;
- «Токоферол» (витамин Е) — жирорастворимый витамин с выраженным антиоксидантным действием, нормализует структуру и проницаемость биологических мембран, оказывает положительное действие на интенсивность клеточного дыхания, активизирует ферменты и коферменты, содержащие сульфгидрильные группы;
- витамин С участвует во многих окислительно-восстановительных процессах благодаря способности обратимо окисляться в дегидроаскорбиновую кислоту. Кроме того, он действует как переносчик электронов на уровнях 3 и 4 дыхательной цепи.
Однако объективного клинического подтверждения длительного улучшения зрительных функций и замедления процесса заболевания при динамическом наблюдении не представлено.
Поиски патогенетически обусловленной терапии продолжаются.
Хирургическое лечение птоза и косоглазия может привести к осложнениям, авторы наблюдали появление стойкой диплопии после удаления катаракты у больных с митохондриальной патологией.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0