Роговица (Часть 2) | Детская офтальмология


Описание
Эктодермальная дисплазияОчень редкий синдром, наследуемый как Х-сцепленное расстройство аутосомно-рецессивному типу. Характеризуется патологией желез внешней секреции, истончением или отсутствием волос, изменениями зубов и ногтей (рис. 11.13).

Описаны две основных формы заболевания гидротическая и гипогидротическая. Патология роговицы при этом расстройстве характеризуется помутнениями, формированием паннуса, а также развитием синдрома «сухих» глаз с сопутствующими вторичными изменениями, включая инфекционный кератит.
Врожденный буллезный эпидермолиз
Дистрофическая форма этого заболевания проявляется дефектом соединения конъюнктивы с дермоэпидермальными тканями и нарушением контакта эпителия роговицы с базальной мембраной.
Патология роговицы при данном расстройстве развивается относительно редко, но возникая, она имеет следующие проявления:
- расширение лимба;
- сетчатые помутнения роговицы:
- симблефарои (рис. 11.14).

Ихтиоз
Ихтиоз объединяет группу расстройств, проявляющихся шелушенном. При тяжелом клиническом течении ихтиоза больные погибают в связи с присоединением распространенной инфекции кожных покровов, не поддающейся терапии.
Известны следующие формы заболевания:
- Ихтиоз vulgaris аутосомно-доминантное расстройство с отсутствием офтальмологических проявлений.
- Х-сцепленная форма ихтиоза шелушение волосистой части кожи головы, лица, шеи, живота и конечностей; утолщение нервов роговицы и диековидная кератопатия. Могут формироваться поверхностные помутнения роговицы, помутнения задних слоев встречаются реже.
- Ламеллярный и линейный ихтиоз тяжелые формы заболевания с аутосомно-рецессивным типом наследования. Глазные симптомы включают эктроппоп и кератоконъюнктивит, как правило, вторичного характера, обусловленные иссмыканием глазной щели.
- Эпидермолизный гиперкератоз и эритрокератодермия — аутосомно-доминантные расстройства с вовлечением в патологический процесс роговицы (рис. 11.15).
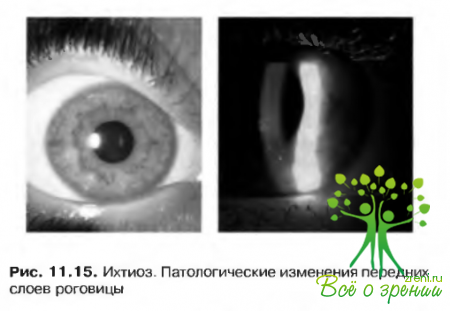
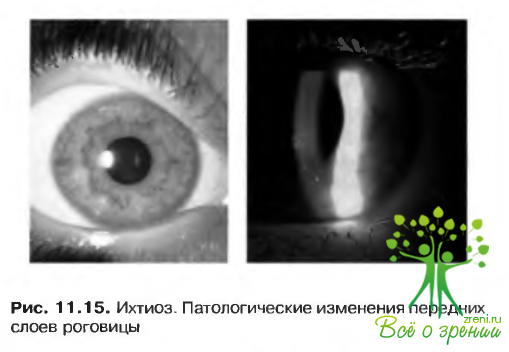
К другим синдромам, характеризующимся ихтиозом, относятся:
- Синдром Сьерепа—Ларссеиа (Sj?gren-Larssen);
- Синдром Нитертона (Netherton);
- Синдром Рефсума:
- Точечная хондродисплазня;
- Синдром IBIDS;
- Синдром KIDS.
Отсутствие и снижение чувствительности роговицы
Расстройства чувствительности роговицы часто провоцируют возникновение кератита. Данная патология известна под названием нейротрофического кератита, хотя основное этиологическое значение имеют такие причины, как исчезновение слезной пленки, сокращение частоты мигательных движений и повторяющаяся травма роговицы, а не нейротрофический фактор.
Возможен вторичный характер расстройства чувствительности роговицы при повреждениях V пары черепно-мозговых нервов в сочетании с:
- herpes zoster,
- травмой;
- внутричерепными опухолями;
- herpes simplex.
- окуло-фацнальным синдромом:
- синдромом Гольдспара (Goldenbar);
- проказой;
- отравлением сероуглеродом и сероводородом;
- синдромом Рапли-Дся;
- синдромом MURCS (рис. 11.16).

При рецидивирующих эрозиях и низкой эффективности проводимого лечения повышается риск присоединения вторичной инфекции. Особенно трудны для коррекции случаи с сопутствующим лагофтальмом и патологией слезного аппарата. Именно это сочетание имеет место при синдроме Райли-Дея (рис. 11.17), и некоторых опухолях мозга.
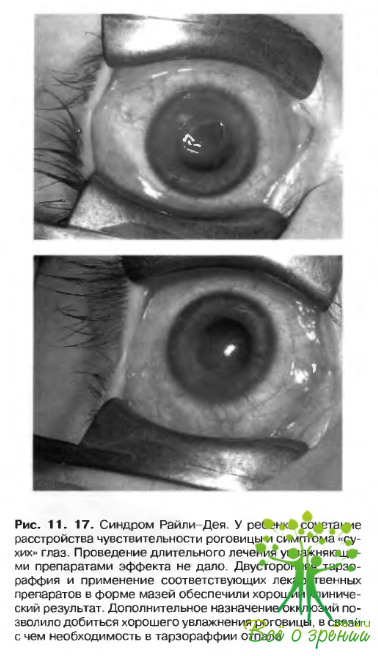
Лечение включает применение искусственной слезы и соответствующих лекарственных препаратов в форме мазей. Периодически, с целью профилактики вторичной инфекции, назначают инстилляции антибиотиков. При сопутствующем лагофтальме обеспечивают смыкание век в ночное время или производят тарзораффню.
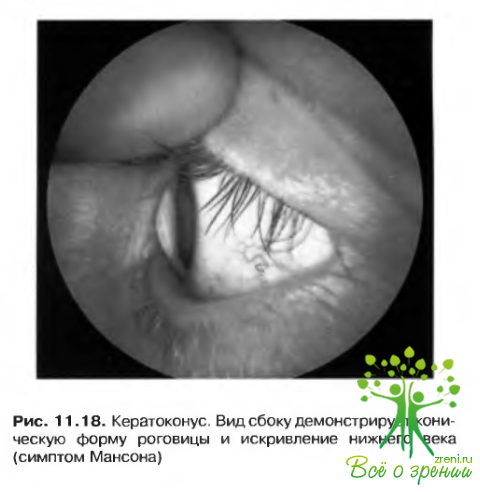
Кератоконус
Кератоконус развивается в связи с дистрофическим растяжением роговицы, приводящим к истончению ее центральных и парацентральных отделов. Заболевание обычно возникает па втором десятилетни жизни (рис. 11.18). Этиология кератоконуса неизвестна, хотя существуют предположения о важной роли травмы в происхождении заболевания. Значение наследственного фактора не определено, хотя у некоторых больных четко прослеживается семейный анамнез. Большинство случаев имеет спорадический характер.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Клинические проявления кератоконуса первоначально связаны с его влиянием па остроту зрения. Истончение роговицы приводит к развитию неправильного астигматизма, что обосновывает целесообразность назначения контактных линз (рис. 11.19).

По мере прогрессировать заболевания появляются разрывы деснеметовой мембраны, связанные с гидратацией и приводящие к острому наводнению роговицы (рис. 11.20). При этом состоянии затуманивание зрения, обусловленное отеком роговины, сопровождается сильной болью.
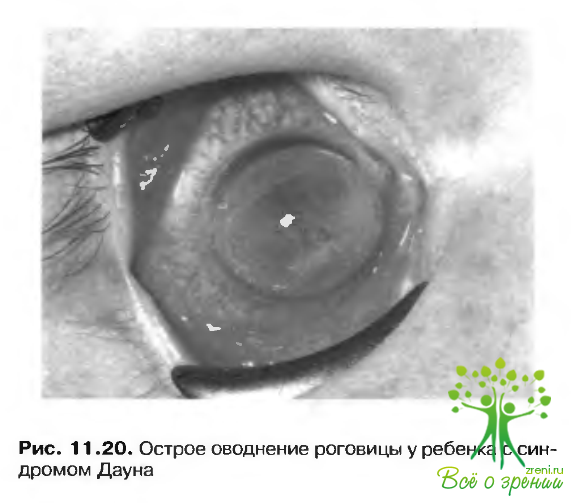
Процесс прекращается спонтанно, оставляя после себя разнообразные рубцовые изменения. Хирургическое лечение в острой стадии не показано.
Лечение заболевания, как правило, консервативное, включающее ношение контактных линз. В тяжелых случаях показана сквозная кератопластика.
Кератоконус может сопутствовать следующим состояниям:
- синдрому Апера (Apert);
- атоппи;
- брахидактилии;
- синдрому Крузона;
- синдрому Элерса-Данто (Ehlers-Danlos);
- синдрому Лоренса-Муна-Бпдля (Laurence- Moon-Bictll);
- синдрому Марфапа:
- пролапсу митрального клапана;
- синдрому Нупапа (Noonan);
- несовершенному остеогепезу;
- синдрому Рейно (Raynaud);
- синдактилии;
- пигментной экземе;
- врожденному амаврозу Лебсра (Leber) (и другим врожденным палочко-колбочковым дистрофиям).
Кератоглобус
При кератоглобусе истончение роговицы локализуется в ее центре. Кератоглобус может сочетаться с кератоконусом, а может возникать как изолированная патология.
В ряде случаев кератоглобус сопутствует таким расстройствам, как:
- голубые склеры;
- разболтанность суставов, глухота, пятнистость зубов;
- синдром Рубинштенна-Тейби (Rubinstein- Taybi) (рис. 11.21).

Метаболические расстройства с поражением роговицы
При метаболических заболеваниях в патологический процесс может вовлекаться любой из слоев роговицы.
Патология эпителия
Эпителии роговицы может прокрашиваться токсическими веществами или вовлекаться в патологический процесс при некоторых расстройствах обмена.
- Хлорохии дифосфат и гидроксихлорин сульфат — кольцевидное помутнение.
- Амподарон — характерные завитки, нежные точечные помутнения (рис. 11.22).

Субэпителиальная патология
Это субэпителиальные линейные помутнения, имеющие радиальное направление и расположенные в верхней половине роговицы. Частые спутники эптеропатического акродерматита. Для этих изменений характерна мутовчатая форма. Процесс может осложниться кератомаляцией.
Симптомы общей патологии включают дистрофию ногтей, расстройства пищеварительной системы, обусловливающие диарею и замедленный рост ребенка.
В качестве лечебного мероприятия назначают диету, обогащенную цинком.
Строма
При цистинозе происходит нарушение транспортной функции лизосом и отложение в них цистеина. Кристаллы цистеина, накапливаясь в передних отделах стромы, вызывают утолщение роговицы, расстройство ее чувствительности, развитие поверхностной точечной кератопатии и рецидивирующих эрозий (рис. 11.23).

Сопутствующая цистинозу патология включает задержку развития, заболевание почек, депигментацию волос и кожных покровов, пигментную ретинопатию. Возникая в раннем детстве, цистиноз приводит к почечной недостаточности и ранней смерти. Развиваясь у взрослых, заболевание не поражает почки и ограничивается роговичными проявлениями. Дебютируя у подростков, цистиноз по клиническим проявлениям напоминает инфантильную форму, но с отсутствием задержки развития и изменений кожных покровов.
Высокоэффективно назначение цистеамина. При значительном снижении зрения показана сквозная Keparoi LiacTiка.
Десцеметовая мембрана
Болезнь Вильсона (Wilson) представляет собой наследственное заболевание с расстройством метаболизма меди. При этой патологии уровень содержания белка, транспортирующего медь, снижен в сыворотке крови, по повышен в тканях. По-видимому, патологический ген подобен гену Менкеса и локализован в зоне 3ql4.3. Расстройство часто проявляется окрашиванием десцеметовой мембраны на периферии роговицы, особенно интенсивным на 12 и 6 часах (рис. 11.24).

Памятка 11.4.
Отложение кристаллов в роговице
Отложение кристаллов в роговице встречается при следующих состояниях:
- цистиноз
- кристаллическая дистрофия роговицы (дистрофия Шнидера);
- дефицит лецитин-холестерол-ацилтрансферазы;
- кристаллы мочевой кислоты;
- гранулярная дистрофия и краевая дистрофия Биетти;
- множественная миелома; моноклональная гаммапатия;
- отложение кальция;
- синдром отложения кристаллов в роговице, миопатия, нефропатия;
- тирозинемия II типа.
На ранних стадиях заболевания этот симптом может быть установлен лишь с помощью гонноскопии.
Сопутствующие изменения включают:
- дегенерацию базального ганглия с появлением тремора;
- хореоатетоз;
- изменения со стороны нервной системы и психики;
- прокрашивание почечных канальцев с появлением аминоацидурии;
- узловая форма цирроза.
Иногда при болезни Вильсона развивается катаракта, напоминающая цветок подсолнечника. Лечение пенициламином не всегда обеспечивает эффект, может потребоваться трансплантация печени.
Лентовидная кератопатия
Лентовидная кератопатия является результатом пропитывания боуменовой мембраны кальцием. Первопричиной выступает, как правило, хронический воспалительный процесс или системное заболевание. Отложение кальция первоначально локализуется в пределах глазной щели.
Лентовидная кератопатия частый спутник таких общих расстройств, как саркопдоз, паратиреоидная патология и множественная миелома.
Во многих случаях лентовидная кератопатия является спутником таких хронических воспалительных процессов, как ювенильный ревматоидный артрит, болезнь Стилла (Still) и саркопдоз (рис. 11.25).

При данной патологии эффективны инстилляции хелатных препаратов. В некоторых случаях лентовидная кератопатия развивается на конечных стадиях фтизиса глазного яблока.
Дефицит лецитин-холестерол- ацилтрансферазы
Заболевание наследуется по аутосомно-рецессивному типу. В гомозиготных состояниях и у недоношенных возникает центральное нежное помутнение роговицы, а в гетерозиготных — формируется arcus senilis. Сопутствующие общие заболевания включают почечную недостаточность, анемию п гиперлинндемию.
Дугообразная дегенерация роговицы
Линоидные Дуги роговицы развиваются в связи с отложением в периферических отделах стромы фосфолипидов, липопротеидов низкой плотности и триглнцеридов.
Дуга чаще формируется у лиц молодого возраста и сопутствует следующим заболеваниям:
- семейная гиперхолнстеролемня (синдром Фредериктона [Friederickson|, тип II) (рис. 11.26);

- семенная гпнерлипонротепнемпя (тип III);
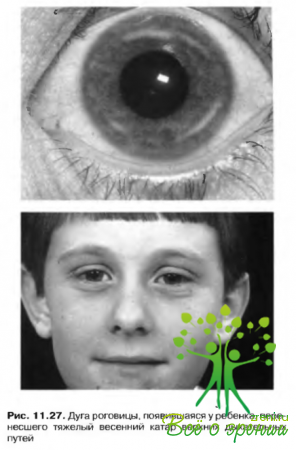
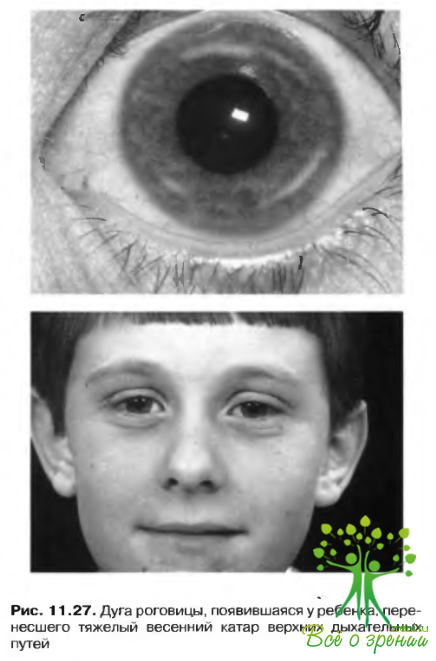
- патологические процессы, сопряженные с заболеваниям роговицы, такие как весенняя кератопатия пли herpes simple.v (рис. 11.27).
- первичная липоидная дегенерация роговицы.

Формирование дуги может встречаться и у здоровых людей при нормальном уровне липидов в плазме крови.
Гифема и прокрашивание роговицы кровью
Прокрашивание роговицы кровью — серьезное осложнение гифемы. При наличии гифемы единственными прогностическими признаками возможного прокрашивания роговицы кровью являются повышенный уровень внутриглазного давления и изменения состояния роговицы. Особенно неблагоприятными факторами в этом отношении выступают повторные кровоизлияния в послеоперационном периоде. Ребенку с гифемой нельзя назначать лекарственные препараты, влияющие на состояние свертывающей системы крови. Прокрашивание роговицы кровью способно понизить остроту зрения и в последующем привести к развитию амблиопии. Иногда для устранения дефекта требуется проведение сквозной кератопластики.
Памятка 11.5.
Голубые склеры
Наследственное расстройство, при котором дефект мезодермальных тканей обусловливает голубоватую окраску склеры, возможно связанную с ее истончением.
Встречается при следующих заболеваниях:
- несовершенный остеогенез;
- синдром Элерса-Данло;
- синдром Халлерманна-Штрайффа (Hallermann- Strieff);
- синдром Марфана;
- истончение роговиц;
- эктодермальная дисплазия.
В младенческом возрасте голубоватая окраска склер встречается у многих совершенно здоровых детей.
Множественная эндокринная неоплазия (МЭН)
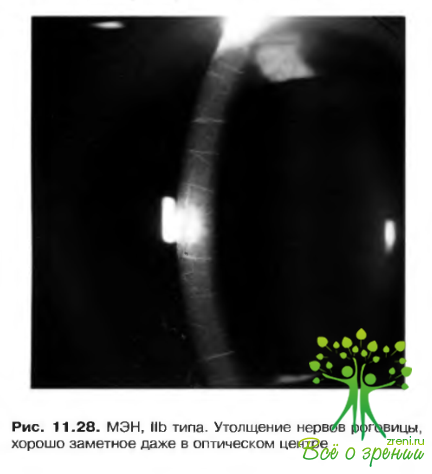
Существуют три основных синдрома, которые включают разные новообразовании в различных органах внутренней секреции у детей. В офтальмологии наибольшее значение имеет МЭН IIb типа. Для больных, страдающих этим расстройством, патогномоничен марфаноподобный фенотип, полные губы, узелковые невромы на кончике и краях языка и век, конская стопа, запоры и перонсальная мышечная атрофия.
Характерным диагностическим признаком является приподнятость нервов роговицы на фойе отсутствия сопутствующих изменений роговицы. В связи с высокой частотой возникновения медуллярной карциномы щитовидной железы при МЭН IIb типа, больным детям в некоторых случаях рекомендуют профилактическую тиреоидэктомию. Заболевание может сопровождать развитие фейохромонитомы (рис. 11.28).
Дистрофии роговицы
Дистрофии роговицы, обычно представляют собой двусторонние и симметричные расстройства наследственного характера. Различают дистрофии роговицы и дегенерации, не имеющие генетической основы и развивающиеся на фоне старения или предшествующего воспаления роговицы.
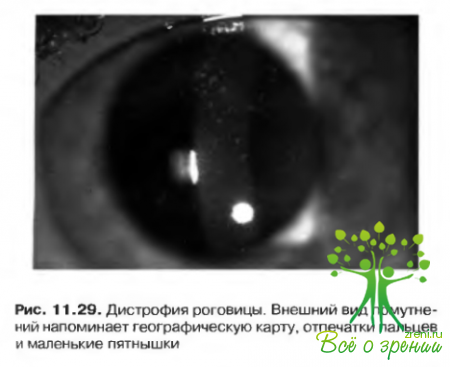
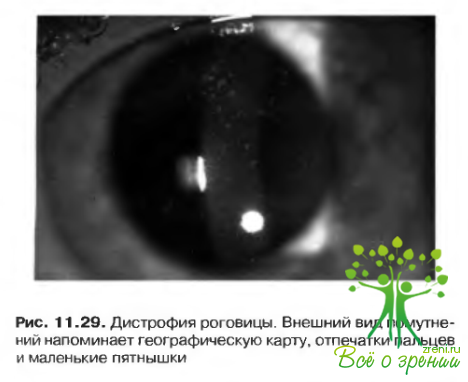
- Изменения роговицы, по внешнему виду напоминающие географическую карту и отпечатки пальцев; пятнышки; микрокисты Когана, дистрофия базальной мембраны. Напоминающие отпечатки пальцев помутнения, локализующиеся под эпителиальным слоем. Острота зрения снижается редко. Расстройство наследуется по аутосомно-доминантному типу и не связано с общей патологией. Единственным значимым осложнением заболевания являются рецидивирующие эрозии (рис. 11.29).
- Ювенилъная эпителиальная дистрофия; дистрофия Месманна-Уилке (Meesman-Wilke). Аутосомно-доминантный тип наследования с неполной пенетрантностью. Основным клиническим проявлением является формирование крошечных пузырьков в эпителиальном слое. Раздражение глазного яблока и светобоязнь обусловлены рецидивирующими эрозиями. Зрение снижается редко. С лечебной целью могут быть назначены контактные линзы.
- Дистрофия Раиса-Бюклера (Reis-Buckleib). Случаи, дебютирующие в раннем детстве, характеризуются аутосомно-доминантным типом наследования. Клинические проявления включают периодически появляющуюся светобоязнь, боль и покраснение глазного яблока. Микроскопические выпячивания связаны с отложением патологических веществ на уровне мембраны. Чувствительность роговицы обычно страдает. Снижение зрения возникает на третьем или четвертом десятилетии жизни. Может потребоваться кератопластика (рис. 11.30).

- Узелковая дистрофия; Греноува (Сгопняс) тип I. Тип наследования аутосомно-доминантных. Характерна постоянная экспрессивность во всех поколениях. Заболевание связано с патологию хромосомы. Наблюдается образование мелких помутнений в виде узелков, первоначально располагающихся над оболочкой, а в дальнейшем захватывающих строму роговицы. Острота зрения длительное время остается нормальной. На пятом-шестом десятилетии жизни может потребоваться сквозная кератопластика.
- Решетчатая дистрофия; дистрофия Гааба— Вибера-Диммера (Haab-Biber- Dimmer). Заболевание наследуется по аутосомно-доминантному типу п связано с патологией 5q хромосомы. Часто имеет асимметричный характер. Связана с отложением в роговице амилоидных веществ. Типичная решетчатая форма заболевания иногда проявляется лишь во взрослом возрасте. Может потребоваться сквозная кератопластика. В некоторых случаях эта патология роговицы возникает па фоне общего амплон-доза или прогрессирующего пареза внутричерепных и периферических нервов.
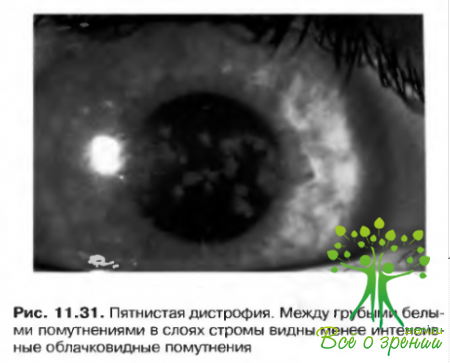
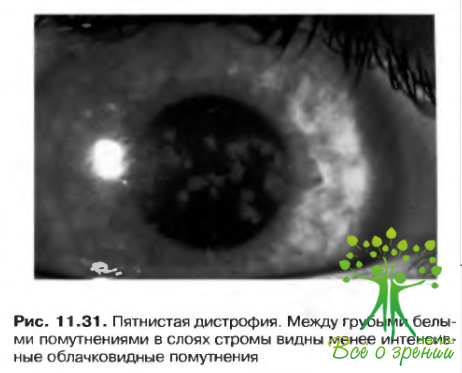
- Пятнистая дистрофия: Греноува тип II. Аутосомно-рецессивный характер наследования. Редко проявляется в детские годы. По мере прогрессировать заболевания возникает утолщение роговицы и нежное помутнение ее с громы. Необходимость в сквозной кератопластике может возникнуть лишь на отдаленных этапах развития патологии (рис. 11.31).
- Центральная кристаллическая дистрофия Шнидера. Аутосомно-доминантный тип наследования с различной экспрессивностью. Характеризуется образованием диековидного помутнения в центре роговицы с включениями пли без включения кристаллов холестерина. Патологии сопутствует липоидная дуга роговицы. Во взрослом возрасте формируется диффузное помутнение в слоях стромы, в связи с чем показано проведение сквозной кератопластики.
- Дермолондриалышя дистрофия роговицы. Редкое заболевание, характеризующееся дистрофическими изменениями передних слоев роговицы в центре. Сочетается с катарактой, деформацией лимба и узелковым поражением кожных покровов.
- Задняя полиморфная дистрофия Шлихтинга. Заболевание наследуется по аутосомно-доминантному типу. Характеризуется появлением асимметричных, медленно прогрессирующих кольцевидных помутнений, локализующихся в глубоких слоях роговицы на уровне десцемстовой мембраны. Патология проявляется уже в детском возрасте. Может сопровождаться изменением глубины передней камеры и развитием глаукомы (рис. 11.32).

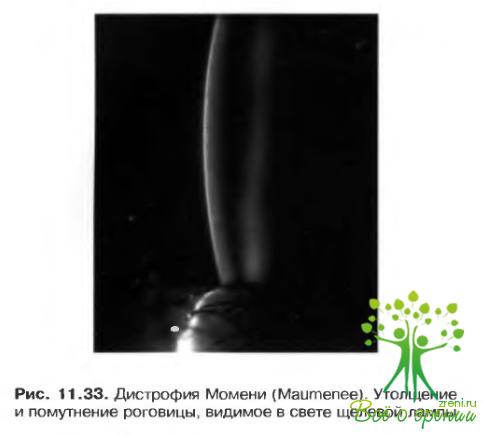
- Дистрофия Моменн (Маитепее). Наследуется по аутосомно-доминантном или аутосомно-рецессивному типу. Дебютирует с рождения. Диффузное аваскулярное помутнение голубовато-белого цвета, напоминающее матовое стекло. Постепенно развивается прогрессирующее утолщение роговицы. С течением времени помутнение способно к самопроизвольному рассасыванию. Необходимость в сквозной кератопластике обычно не возникает (рис. 11.33).


Памятка 11.6.
Разрастание нервов роговицы
- МЭН IIb типа.
- Дистрофические процессы — дистрофия Фукса (Fuchs), кератоконус.
- Воспалительные заболевания — проказа.
- Болезнь Рефсума (Refsum).
- Ихтиоз.
- Неирофиброматоз.
- Буфтальм

Комментариев 0