Болезни крови и кроветворных органов


Содержание:
Описание
↑ Анемии
Анемией, или малокровием, называют состояние, которое характеризуется уменьшением количества эритроцитов и снижением содержания гемоглобина в единице объема крови. В ряде случаев при анемии обнаруживаются и качественные изменения эритроцитов.
При анемии в результате нарушения транспортной функции развиваются явления гипоксии, признаками которой являются одышка, тахикардия, неприятные ощущения в области сердца, головокружение, слабость, быстрая утомляемость, бледность кожных покровов и видимых слизистых оболочек. Выраженность указанных симптомов зависит от степени анемии и быстроты ее развития. При глубокой анемии наряду с указанными симптомами возникают и нарушения зрения.
По цветовому показателю анемии делят на гипохромные, нормохромные и гиперхромные. По величине среднего диаметра эритроцитов анемии разделяют на микроцитарные, нормоцитарные и макроцитарные. По характеру регенерации различают анемии регенераторные, гипорегенераторные, гипо- и апластические, диспластические или дизэритропоэтические.
В настоящее время общепринятой классификацией, построенной по патогенетическому принципу с учетом этиологических и важнейших клинико-морфологических форм, является классификация, предложенная Г. А. Алексеевым (1970).
I. Анемии вследствие кровопотерь (постгеморрагические).
II. Анемии вследствие нарушенного кровообращения:
A. Железодефицитные анемии («хлоранемии»).
Б. Железонасыщенные, сидероахрестические анемии.
B. В12 (фолиево)-дефицитные, «пернициозные» анемии:
1. Экзогенная недостаточность витамина В12 (фолиевой кислоты).
2. Эндогенная недостаточность витамина B12 (фолиевой кислоты):
а) нарушенная ассимиляция пищевого витамина В12 вследствие выпадения секреции желудочного мукопротеина;
б) нарушенная ассимиляция витамина B12 (фолиевой кислоты) в кишечнике;
в) повышенное расходование витамина B12 (фолиевой кислоты).
Г. В12 (фолиево)-«ахрестические» анемии.
Д. Гипоапластические анемии:
1. Вследствие воздействия экзогенных факторов.
2. Вследствие эндогенной аплазии костного мозга.
Е. Метапластические анемии.
III. Анемии вследствие повышенного кроворазрушения (гемолитические):
А. Анемии, обусловленные экзоэритроцитарными гемолитическими факторами.
Б. Анемии, обусловленные эндоэритроцитарными факторами:
1. Эритроцитопатии.
2. Энзимопении:
а) дефицит глюкозо-6-фосфат-дегидрогеназы;
б) дефицит пируваткиназы;
в) дефицит глутатион-редуктазы.
3. Гемоглобинопатии.
Ниже описываются характерные особенности отдельных форм анемий, при которых наиболее часто встречаются глазные симптомы.
Острая постгеморрагическая анемия развивается вследствие острой однократной и повторной кровопотери от травм, кровотечений из желудочно-кишечного тракта, при внематочной беременности, маточных кровотечениях и др. Симптомы болезни патогенетически связаны с уменьшением массы циркулирующей крови и кислородной недостаточностью. Клиническая картина в первые моменты после массивной кровопотери укладывается в клинику пост-геморрагического шока или коллапса: бледность кожных покровов, обморочное состояние, головокружение, холодный пот, частый нитевидный пульс, иногда рвота, судороги. В дальнейшем по мере улучшения общего состояния и стабилизации артериального давления в клинической картине начинают преобладать симптомы малокровия и гипоксии. Именно в этом периоде наиболее часто выявляются признаки нарушения зрения вплоть до полного амавроза, так как специфические элементы сетчатки очень чувствительны к анемии.
При хронических гипохромных железодефицитных анемиях, включая ранний и поздний хлороз, симптоматических железодефицитных анемиях (хронический энтерит, агастрическая хлоранемия, грыжа пищеводного отверстия диафрагмы, злокачественные новообразования, хронические инфекции), а также хронически протекающих гипохромных мегалобластных анемиях (пернициозyые анемии различного генеза — анемия Аддисона—Бирмера, глистная, спруанемия, целиакия и др.) выраженность глазных симптомов зависит от степени анемии, которая, правда, индивидуально варьирует в широких пределах. Особенно часто изменения на глазном дне возникают при концентрации гемоглобина ниже 5 г% и реже 7 г% [Merin S., Preud М., 1968].
Глазное дно при анемии выглядит бледным. Этот симптом не всегда можно оценить из-за различий в пигментации сетчатки и хориоидеи. Легче обнаруживается деколорация диска зрительного нерва и сосудов сетчатки. При этом артериальные сосуды имеют тенденцию расширяться и приближаться по калибру к аналогичным венозным веточкам.Множественные кровоизлияния в сетчатку — наиболее характерный симптом ретинопатии при анемиях (рис. 34).
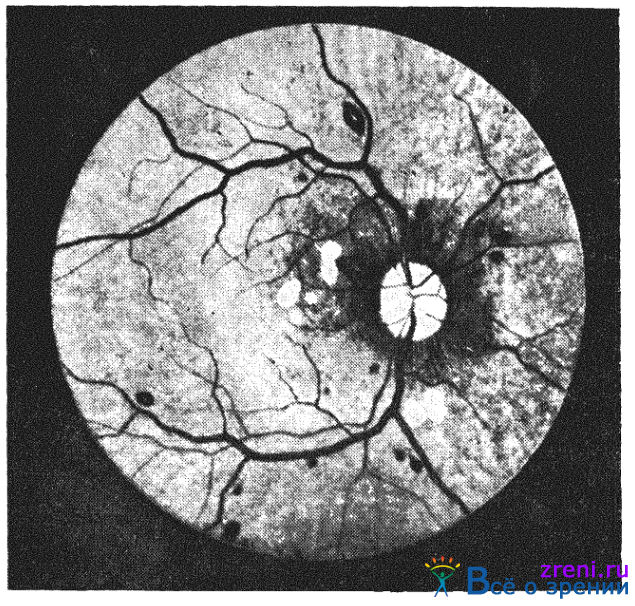
Рис. 34. Глазное дно при пернициозной анемии.
Причина кровоизлияний не вполне ясна. По-видимому, недостаток кислорода обусловливает повышенную проницаемость капилляров. При пернициозной анемии имеет значение и сопутствующая тромбоцитопения.
Полосчатые или в форме пламени геморрагии расположены в слое нервных волокон. Они могут локализоваться в любом отделе сетчатки, но их нет в желтом пятне. Поэтому острота зрения обычно сохраняется. Иногда в экстравазатах виден белый центр. Этот симптом чаще наблюдается при пернициозной анемии. В некоторых случаях ишемия может быть причиной отека диска зрительного нерва и прилежащей сетчатки. Обычно отек выражен нерезко, но описаны и случаи застойного диска [Kolker А., 1966]. Кроме отека в слое нервных волокон могут быть небольшие белые очаги, которые состоят из фибрина и обычно хорошо рассасываются при улучшении состояния больного.
Значительно более тяжелые изменения сетчатки наблюдаются при серповидноклеточной (дрепаноцитарной) анемии. Это заболевание относится к наследственно-семейной гемолитической анемии, характерной особенностью которой является свойство эритроцитов принимать серповидную форму — этим заболеванием страдаютпреимущественно негры и редко — лица белой расы. В Советском Союзе описаны единичные случаи.
Заболевание относится к группе гемоглобинопатий с врожденной неполноценностью эритроцитов, в частности с наличием в них патологического глобулина.
Заболевание проявляется уже в детском возрасте и характеризуется хроническим течением с частыми обострениями в виде гемолитических арегенераторных, тромботических и секвестральных кризов.
При гемолитических кризах содержание эритроцитов может в течение короткого периода снижаться до 1—2 млн. ,в 1 мм3 крови. Криз сопровождается развитием желтухи и абдоминального синдрома. Арегенераторные кризы — это временное, функциональное истощение костно-мозгового кроветворения. Тромботические или болевые кризы, которые иногда доминируют в проявлениях болезни, возникают на почве генерализованных тромбозов мелких сосудов, особенно брюшной полости и конечностей. Секвестральные кризы — это состояния, напоминающие шок с внезапным развитием анемии без гемолиза [Токарев Ю. Н., 1966].
Как и при других врожденных гемолитических анемиях, больные серповидноклеточной анемией инфантильны, страдают гипо- гонадизмом, имеют башенный череп и др. При этом заболевании: особенно отчетливо выражен костно-суставной синдром (дактиллиты, боли, деформации, некрозы суставных головок и костей). На голенях часто развиваются хронические язвы. Селезенка и печень увеличены. Тромбозы и эмболии — весьма характерный признак. Поражения сетчатки локализуются преимущественно в экваториальной и периферической зонах и проходят 5 стадий [Goldberg М., 1971]. I стадия характеризуется периферической артериолярной обструкцией, II — появлением артериовенозных анастомозов. В III стадии развивается неоваскулярная и фиброзная пролиферация, которая приводит в IV стадии к кровоизлияниям в стекловидное тело. В конечном итоге (V стадия) развивается отслойка сетчатки.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
↑ Лейкозы
Под лейкозами понимают неопластические заболевания, опухолевая масса которых состоит из клеток крови или, что, по-видимому, более точно, из клеток, сходных по своему виду с клетками крови.
Некоторыми учеными опухоли крови подразделяются на гемобластомы и гематосаркомы на том основании, что в отдельных случаях костный мозг может быть повсеместно заселен этими опухолевыми клетками, а в других случаях их разрастание осуществляется экстрамедуллярно. На наш взгляд, такое подразделение часто осуществить очень сложно, так как опухолевые разрастания лейкемических клеток могут иметь экстрамедуллярную локализацию и у больных, у которых заболевание началось с поражения костного мозга. И, наоборот, в ряде случаев гематосарком впоследствии может происходить вовлечение в процесс костного мозга, и клиницисты вынуждены в этих случаях говорить о лейкемизации процесса. На наш взгляд, более правильным все опухоли кроветворной ткани объединить под названием «лейкозы», так как опухолевая неопластическая природа этих заболеваний, подчеркиваемая в названиях «гемобластозы» или «гематосаркоматозы», ни у кого практически не вызывает сомнений.
Этиология лейкозов не может считаться окончательно выясненной, что касается, впрочем, и других опухолей в равной мере. Однако в настоящее время можно считать установленным, что такие факторы, как вирус, ионизирующая радиация, определенные химические субстанции, в том числе и некоторые лекарственные вещества типа левомицетина, бутадиона и цитостатиков, способны оказывать определенное стимулирующее влияние на возникновение данных заболеваний. О роли наследственных факторов в возникновении лейкозов также имеются вполне обоснованные мнения. Они подтверждаются случаями возникновения однотипных лейкозов у однояйцевых близнецов, большой подверженности развитию лейкозов у больных с наследственными нарушениями генетического аппарата — болезнью Дауна, синдромами Тернера,.. Клайнфелтера и т. п. При этом отмечено, что определенные виды лейкозов имеют тенденцию сочетаться с определенными типами генетических нарушений. Необходимо иметь в виду, что современные научные данные весьма убедительны в пользу выдвигавшегося ранее предположения о происхождении всей лейкемической массы из одной мутировавшей клетки, вышедшей из-под контрола организма больного. Таковыми являются наличие кольцевой хромосомы в опухолевых клетках больных острыми лейкозами, развившимися у лиц, леченных радиоактивным фосфором, резкое увеличение содержания однотипного по физико-химическим свойствам протеина у больных парапротеинемическими гемобластозами,. филадельфийская хромосома у больных хроническим миелолейкозом.
В клинической практике лейкозы принято подразделять в зависимости от типа клетки, составляющей основу опухолевой массы. Те лейкозы, которые протекают с пролиферацией клеток малодифференцированных и не способных к дальнейшей дифференциации, обычно без лечения весьма злокачественны и называются острыми. Лейкозы, опухолевую массу которых составляют клетки дифференцирующиеся и зрелые, обычно имеют относительно доброкачественное течение и носят название хронических лейкозов.
Острые и хронические лейкозы в свою очередь подразделяются в зависимости от того, какая клетка составляет субстрат опухоли. В настоящее время описаны лейкозы, развивающиеся из клеток всех ростков кроветворения — эритроидного, тромбоцитарного,. гранулоцитарного и агранулоцитарного типа. При этом различают острые лейкозы миело-, моно-, мегакарио-, эритро- и плазмобластного типов. Так как дифференциация острых лейкозов осуществляется лишь на основании цитохимических методов исследования, а цитохимические способы идентификации клеток осуществляются с помощью эмпирически отобранного набора методов, появились сообщения о существовании такой формы острого лейкоза, как недифференцируемый. Происхождение последнего, по-видимому, можно отнести за счет пролиферации клеток, происходящих из более ранних, неотдифференцированных клеток кроветворения. Среди хронических лейкозов выделены и продолжают выделяться формы лейкозов, в основе которых лежит пролиферация любой зрелой клетки крови. Здесь и хронический лимфолейкоз, хронический миелолейкоз, хронический моноцитарный лейкоз, хронический мегакариоцитарный лейкоз, эритромиелоз, эритремия, плазмоцитома, хронический базофильноклеточный лейкоз; имеются сообщения и о наличии хронического эозинофильного лейкоза.
При современном уровне медицинской науки, позволяющей различать тончайшие детали клеток, производятся подразделения в рамках как будто бы давно устоявшихся форм лейкозов. Так, среди группы больных с хроническим лимфолейкозом в настоящее время уже выделяют группы лиц, страдающих от пролиферации как Т-, так и В-лимфоцитов, а среди больных хроническим миелолейкозом различают группы с пролиферацией клеток, имеющих филадельфийскую хромосому и не имеющих ее. Не исключена возможность, что идентификация лейкозов будет продолжаться и в дальнейшем, и это позволит осуществлять более специфическое и более действенное лечение больных.
На основании изложенного выше говорить о диагностике как самого лейкоза, так и о его конкретной форме довольно несложно. Диагностика этого заболевания осуществляется при констатации гиперплазии кроветворной ткани, которая может иметь место как в периферической крови, так и в костном мозге. При этом у отдельных лиц гиперплазия лейкемических клеток имеет место только в костном мозге, а в периферической крови эти клетки появляются лишь на более поздних стадиях заболевания. В связи с этим исследования костномозгового кроветворения при помощи анализа данных стернального пунктата, а иногда и структуры костной ткани с помощью трепанобиопсии должны производиться в процессе диагностики. Применение же цитохимических и цитогенетических методов исследования обычно приводит лишь к уточнению варианта лейкоза.
Возможность существования лейкемоидных реакций, т. е. таких разрастаний кроветворной ткани, возникающих в ответ на наличие в организме больного какого-то фактора, активирующего кроветворение, заставляет иногда проводить специальные исследования, исключающие наличие этих причин гиперплазии кроветворной ткани.
Клиническая картина лейкозов весьма разнообразна. При этом разнообразие клинических проявлений имеется у больного как острыми, так и хроническими лейкозами. Предсказать дальнейшее клиническое течение, клинические проявления лейкоза у отдельного больного, по-видимому, не решится ни один опытный клиницист. Сделать это практически невозможно в связи с тем, что высокая морфодинамичность и почти повсеместное возможное распространение лейкемической ткани в организме больного могут демонстрировать самую разнообразую симптоматику, симулируя, особенно в начальных стадиях, заболевания самого различного характера. Примером этому может служить работа одного из основоположников отечественной гематологии акад. И. А. Кассирского,, который вместе со своими сотрудниками при анализе первичных диагнозов, с которыми больные поступали в клинику и у которых впоследствии был верифицирован острый лейкоз, обнаружил более 60 различных нозологических форм, среди которых фигурировали сепсис, рак желудка, ревматизм и острая кишечная непроходимость, инфаркт миокарда, ревматоидный артрит, острый менингит и многие другие заболевания.
В то же время о клинике лейкозов говорить можно и довольно просто в связи с тем, что все клинические проявления этих заболеваний можно объединить и понять на основании распознавания главных синдромов, которые обычно с тем или иным преобладанием в зависимости от типа лейкоза имеют место в клинической картине заболевания. Среди этих синдромов наиболее часто встречаются следующие: 1) общетоксический синдром (или интоксикационный) ; проявлением его являются лихорадка, слабость, потливость, похудание, отсутствие аппетита и т. д.; 2) геморрагический синдром. Проявления его чрезвычайно разнообразны, включая меноррагии, кожные кровоизлияния и кровоизлияния в головной мозг; 3) синдром токсико-некротических поражений слизистых оболочек желудочно-кишечного тракта; 4) анемический синдром; 5) синдром опухолевого роста, характеризующийся разрастанием лейкемической ткани в организме. Сюда же нужно относить увеличение лимфатических узлов, печени, селезенки, нарушения функций внутренних органов за счет их сдавливания или же нарушения целостности разрастающейся лейкемической тканью.
Кроме проявлений этих синдромов, характерных для всех лейкозов, отдельные виды лейкозов, в частности парапротеинемические гемобластозы (плазмоцитома, болезнь Вальденштрема, болезни тяжелых и легких цепей), эритремия, имеют в клинической картине ряд особенностей, которые будут описаны в отдельных разделах. Особую окраску клинической картине лейкозов (лимфатического типа) иногда могут придавать аутоиммунные реакции, проявляющиеся гемолитической анемией, лихорадкой, кожными изменениями и т. д.
Не останавливаясь на внешних проявлениях каждого из перечисленных выше синдромов, хотелось бы отметить, что за последние годы в клинической картине лейкозов стали отмечаться проявления, которые могут быть объяснены как цитостатической терапией, так и удлинением срока жизни больных с этой патологией. К ним относятся учащение инфекционных осложнений, которые являются причиной смерти почти 40% больных хроническим лимфолейкозом, учащение неврологической симптоматики (особенно у больных острыми лейкозами, получившими название нейролехь коза), а также частое развитие у больных лейкозами мочекислой нефропатии с явлениями почечнокаменной болезни.
Таким образом, клиника лейкозов может характеризоваться самой разнообразной симптоматикой, которая является следствием разнообразного сочетания указанных выше синдромов. Конечно, при отдельных видах лейкозов можно отметить преобладание того или иного синдрома из перечисленных выше, однако недооценивать возможности подключения любого из них в клинической картпне при любом виде лейкоза не может ни один клиницист.
Говоря о лейкозах, нельзя не упомянуть того большого прогресса, который достигнут современной медициной в лечении этих заболеваний. Ведь именно при этом виде опухолей получены результаты, позволяющие говорить о принципиальном излечении человека от злокачественного неопластического заболевания. Излечение больных острым лимфобластным лейкозом, лимфогранулематозом позволяет надеяться, что эти успехи распространятся и на лечение остальных форм лейкозов.
Острые и хронические формы лейкозов сопровождаются одинаковыми глазными проявлениями, вызванными повышением вязкости крови, гипоксией и лейкемической инфильтрацией ткани. Эти изменения включают в себя образование микроаневризм в сосудах сетчатки, геморрагии, клеточную инфильтрацию хориоидеи, сетчатки, зрительного нерва и периорбитальных структур. Инфильтрация оболочек мозга может привести к параличу экстраокулярных мышц и развитию застойного диска. Описана также инфильтрация век, конъюнктивы, орбитальной клетчатки с развитием экзофтальма.
При офтальмоскопии отмечается бледный фон глазного дна. Ретинальные вены расширены, извиты, и в сетчатке по их ходу нередко видны беловатые полосы, представляющие собой периваскулярную лейкемическую инфильтрацию. Артерии изменены значительно меньше, чем вены.
Величина и форма геморрагий варьирует. Они могут быть глубокими, поверхностными или даже преретинальными. Нередко в центре ретинального кровоизлияния можно видеть белый участок, вызванный аккумуляцией лейкоцитов. В наиболее тяжелых случаях появляются ишемические ватообразные очаги в слое нервных волокон, выраженный отек диска зрительного нерва и перипапиллярной сетчатки и новообразованные ретинальные сосуды.
Изменения на глазном дне при лейкозах встречаются примерно в 70% случаев, особенно часто при острых формах. Тяжесть изменений более или менее коррелирует с тяжестью болезни, и при эффективном лечении основного заболевания улучшается и состояние глазного дна.
↑ Полицитемии
Термин «полицитемия» включает группу заболеваний, которые проявляются увеличением массы эритроцитов в организме, т. е. увеличением их объема на 1 кг массы тела. Число эритроцитов в 1 мм3 крови при полицитемии повышается до 7—10 млн., а содержание гемоглобина до 180—240 г/л. Различают «истинную» полицитемию (эритремия, болезнь Вакеза) и вторичные (симптоматические) эритроцитозы.
Эритремия — первичное миелопролиферативное заболевание кроветворной системы, в основе которого лежит тотальная гиперплазия клеточных элементов костного мозга, особенно его зрительного ростка. Поэтому повышенное содержание лейкоцитов (до 9000—15 000 млн. в 1 мм3 крови) и тромбоцитов (до 1 млн. и более) в крови наряду с более заметным увеличением количества эритроцитов является весьма характерным признаком эритремии. G. F. Stroebe (1951) выделил три гематологических варианта эритремии: 1) без существенного увеличения числа лейкоцитов и Изменений в формуле крови; 2) с умеренным лейкоцитозом, нейтрофплезом и палочкоядерным сдвигом; 3) с высоким лейкоцитозом, нейтрофилезом и сдвигом в формуле крови до миелоцитов. При «истинной» полицитемии обнаруживаются признаки миело фиброза и остеомиелосклероза с миелоидной метаплазией селезенки. Как и при других миелопролиферативных заболеваниях, в сыворотке крови больных полицитемией нередко обнаруживается повышение концентрации щелочной фосфатазы, мочевой кислоты и витамина В12. Клиническая картина истинной полицитемии различна в зависимости от фазы болезни и тяжести течения.
В развернутой, собственно эритремической фазе болезни характерными симптомами являются: 1) изменение окраски кожных покровов и видимых слизистых оболочек; 2) увеличение селезенки и печени; 3) повышение артериального давления; 4) тромбозы и геморрагии.
Кожные покровы изменены у подавляющего большинства больных. Они приобретают красно-цианотичный оттенок. Особенно отчетливо изменяется окраска щек, кончиков ушей, губ и ладоней. Подчеркиваем, что в окраске кожных покровов преобладает красный тон, но не яркий, а вишневый. Подобный оттенок приобретают и видимые слизистые оболочки губ, языка и мягкого неба. Сосуды склер заметно инъецированы (симптом кроличьих глаз). На щеках, губах, кончике носа, особенно у женщин, нередко обнаруживаются телеангиэктазии.
Весьма характерным симптомом эритремии является спленомегалия, что связано с миеломной ее метаплазией и повышенным кровенаполнением. У больных истинной полицитемией обычно увеличена и печень. Увеличение ее размеров связывают также с повышенным кровенаполнением, миелоидной метаплазией, разрастанием соединительной ткани вплоть до развития цирроза или тромбоза внутрипеченочных вен (синдром Бадда—Киари). У ряда больных течение болезни осложняется развитием холелитиаза и хронического холецистогепатита. К развитию этих осложнений приводит свойственная больным эритремией пленохромия желчи.
Почти у половины больных эритремией выявляется гипертония, патогенез которой рассматривается в плане компенсаторной реакции организма в ответ на уменьшение ударного и минутного объема крови, повышение ее вязкости и увеличение периферического сопротивления (А. В. Демидова, Е. М. Щербак). Сочетание повышенного артериального давления с увеличенной селезенкой является кардинальным признаком истинной полицитемии. Если при этом у больного увеличивается масса эритроцитов, то диагноз полицитемии становится несомненным.
Характерна парадоксальная склонность больных полицитемией и к тромбозам (крупных артериальных и венозных сосудов мозга, сердца, печени и селезенки, мелких сосудов кистей и стоп) и к повышенной кровоточивости (из язв желудка и двенадцатиперстной кишки, после экстракции зубов, кожные геморрагии и кровотечения со слизистых оболочек). Причиной кровоточивости при истинной полицитемии является увеличение массы циркулирующей крови с переполнением ею сосудов и паретическим расширением капилляров, а также дефицит плазменных факторов свертывания, в частности фибриногена [Мачабели М. С., 1962], серотонина [Матвеенко JI. А., 1965].
Развитие тромбозов при эритремии связано с повышением вязкости крови, замедлением тока крови, увеличением числа тромбоцитов и эритроцитов, со склеротическим поражением стенок сосудов, общей гиперкоагулабильностью крови.
У больных эритремией нередко поражаются почки (в них развиваются инфаркты вследствие тромбоза сосудов или почечнокаменной болезни как результат нарушения пуринового обмена, что свойственно миелопролиферативным заболеваниям).
Истинная полицитемия характеризуется длительным течением, которое может быть легким, средней тяжести и тяжелым. В развитии болезни выделяют три периода, или фазы. Первая фаза заболевания в течение длительного периода может протекать латентно или с нерезко выраженными клиническими симптомами. На ранних стадиях заболевание нередко принимается за гипертоническую болезнь.
Описанная выше клиническая картина характеризует развернутую вторую, так называемую эритремическую фазу. И в этой фазе течение заболевания может быть разнообразным.
Терминальная фаза характеризуется развитием вторичного лшелофиброза с анемией и исчезновением внешних признаков эритремии или развитием острого гемоцитобластоза, реже — ретикулеза.
В отличие от истинной полицитемии вторичные эритроцитозы являются не самостоятельными нозологическими единицами, а лишь симптомами других заболеваний. Увеличение количества эритроцитов и гемоглобина связано не с пролиферативным процессом в костном мозге, а с его функциональным раздражением (абсолютные эритроцитозы) или со сгущением крови без усиления эритропоэза (относительные эритроцитозы). В приведенной ниже классификации указаны основные виды вторичных эритроцитозов, варианты их течения, основные патогенетические механизмы, лежащие в основе их развития, и конкретные заболевания, сопровождающиеся развитием вторичных эритроцитозов.


Наиболее бросающийся в глаза симптом полицитемии — плетора лица и конъюнктивы. Конъюнктивальные и эписклеральные сосуды, особенно вены, расширены, извитые, насыщенно-красного цвета. Такой же вид имеют сосуды сетчатки (рис. 35).
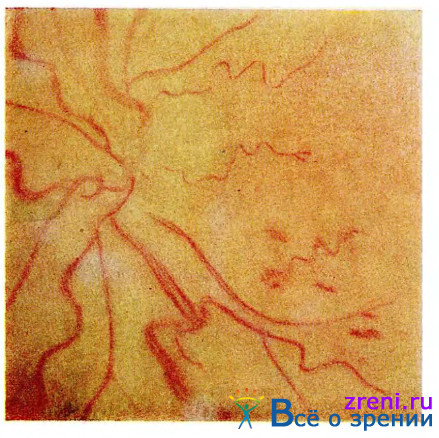
Рис. 35. Глазное дно при полицитемии.
Обращает на себя внимание темно-красный цвет глазного дна. Диск зрительного нерва также необычно красного цвета. Нередко можно видеть более или менее выраженный отек диска зрительного нерва и перипапиллярной сетчатки и единичные геморрагии.
В некоторых случаях развивается окклюзия центральной вены сетчатки. Окклюзия, по-видимому, носит неполный характер. Прогноз в таких случаях обычно благоприятный, во всяком случае значительно лучший, чем при окклюзии центральной вены сетчатки другой этиологии.
↑ Парапротеинемии
В эту группу заболеваний относятся прежде всего миеломная болезнь (плазмоклеточный парапротеинемический ретикулез или болезнь Рустицкого) и макроглобулиновый ретикулолимфоматоз (болезнь Вальденстрема, или макроглобулинемическая пурпура).
Миеломная болезнь является системным заболеванием крови опухолево-гиперпластического типа со злокачественной пролиферацией клеток ретикулоплазматического типа. Это — лейкоз—ретикулоз, в частности плазмоклеточный пара-(или пато-) протеинемический ретикулез.
В зависимости от преобладающего вида клеток выделяют три типа миелом: 1) ретикулоплазмоцитому, 2) плазмобластому и 3) плазмоцитому.
Протеинурия — весьма частый симптом миеломной болезни. С мочой выделяется, как правило, микромолекулярный белок (белок Бенс-Джонса). С протеинурией связано развитие миеломной нефропатии — парапротеинемического нефроза, заканчивающегося обычно смертью при явлениях азотемической уремии.
С высокой концентрацией белка в крови связана и свойственная миеломной болезни высокая вязкость крови.
Болезнь Вальденстрема в настоящее время рассматривается как макроглобулиновый ретикулолимфоматоз, характерной особенностью которого является способность синтезировать макроглобулины: в крови появляются глобулины с молекулярной массой свыше 1 ООО ООО. Заболевают преимущественно люди пожилого возраста. В клинической практике преобладает геморрагический синдром, иногда с чрезвычайно обильными носовыми кровотечениями. Механизм геморрагического синдрома сложен и до конца не уста новлен. Предполагают, что он связан, с одной стороны, с неполноценностью тромбоцитов, взаимодействующих с макроглобулинами, а с другой — с повышенной проницаемостью стенок сосудов вследствие инфильтрации их патологическими белками, высокой вязкости крови и внутрисосудистой агглютинации эритроцитов.
Выделяют преимущественно скелетные формы и скелетно-висцеральные формы заболевания. В патогенетическом плане клиническая картина болезни сводится к двум синдромам, а именно к поражению костей и патологии белков крови. Поражение костей проявляется болями, переломами и развитием опухолей. Особенно часто поражаются позвоночник, кости таза, ребра и череп с развитием соответствующей неврологической симптоматики.
Висцеральная патология проявляется главным образом поражением печени, селезенки, лимфатических узлов и почек. Ее развитие связано как со специфической клеточной инфильтрацией указанных органов, так и с выраженными изменениями белков крови, с накоплением в крови аномального белка — парапротеина, продуцируемого миеломными клетками. При миеломной болезни протеинемия может достигать 12—18 г%.
Ретинопатия при начальных формах миеломной болезни и болезни Вальденстрема отсутствует. У ряда больных глазное дно представляет собой своеобразную картину fundus paraproteinemicus. Характерно расширение ретинальных вен и увеличение их извитости. Артерии также расширяются, но в значительно меньшей степени. Затем проявляются симптом перекреста (передавливание вены под артерией), микроаневризмы, окклюзия мелких вен, геморрагии в сетчатку. В некоторых случаях возникают также ватообразные очаги в слое нервных волокон сетчатки и отек диска зрительного нерва.
Полагают, что указаные изменения в сетчатке связаны как с гиперпарапротеинемией, так и с высокой вязкостью крови. В азотемической стадии болезни развивается характерная для хронических заболеваний почек ретинопатия.
Что касается изменений сосудов сетчатки, то их связь с повышенной вязкостью плазмы крови была продемонстрирована экспериментально [Mausolf F., Mensher J., 1973]. После введения обезьянам в кровь декстрана с высокой относительной массой на глазном дне были выявлены расширенные и извитые ретинальные сосуды, особенно вены, микроаневризмы и геморрагии.
Миеломная болезнь может поражать также кости орбиты, веки, слезную железу, слезный мешок и конъюнктиву, инфильтрировать склеру, радужку, хориоидею, сетчатку и зрительный нерв. Эти поражения, однако, не связары с повышенной вязкостью крови.
↑ Геморрагические диатезы
Под геморрагическими диатезами подразумеваются такие патологические состояния, которые проявляются повышенной кровоточивостью при отсутстви существенных повреждений сосудистой стенки, т. е. кровотечения развиваются в тех ситуациях, когда у других здоровых в этом плане людей их нет.
Важность проблемы геморрагических диатезов весьма велика. Во-первых, это объясняется тем, что количество людей, страдающих повышенной кровоточивостью, в мире превысило шестизначную цифру. Во-вторых, люди, страдающие геморрагическими диатезами, не могут считаться полноценными членами общества, поскольку их потенциальные возможности резко ограничены как анемизацией, часто сопутствующей этой патологии, так и теми видами деятельности, которые предохраняют сосуды больного от различных повреждений.
В-третьих, важность информации о наличии у больных геморрагического диатеза определяется тем, что многие формы этого страдания протекают скрыто или же проявляются слабо, имея моносимптомную клинику. В случае необходимости оперативных вмешательств, даже таких незначительных, как экстракция зуба или тонзиллэктомия, а также при назначении некоторых лекарств, таких, как ацетилсалициловой кислоты, геморрагические диатезы могут оказывать угрозу самой жизни больного.
Патогенез геморрагических диатезов в настоящее время может считаться достаточно хорошо изученным. Как известно, ограничение кровоточивости у здорового человека при повреждении сосудистой стенки осуществляется за счет следующих механизмов: сокращения сосуда в месте его повреждения, оседания на месте повреждения сосуда циркулирующих тромбоцитов и образования ими первичной гемостатической пробки и закрепления ее фибриновой стенкой с формированием окончательной «вторичной» гемостатической пробки. Нарушение любого из этих механизмов приводит к нарушению процесса гемостаза и развитию геморрагического диатеза.
Современные представления о механизмах свертывания крови позволяют нам предложить следующую рабочую классификацию геморрагических диатезов.
КЛАССИФИКАЦИЯ ГЕМОРРАГИЧЕСКИХ ДИАТЕЗОВ
I. Геморрагические диатезы, обусловленные дефектом прокоагулянтов (гемофилии) :
а) недостаточное количество одного или нескольких факторов, участвующих в формировании фибрина;
б) недостаточная активность факторов-прокоагулянтов;
в) наличие ингибиторов отдельных прокоагулянтов в крови больного.
II. Геморрагические диатезы, обусловленные дефектом тромбоцитарного звена гемостаза:
а) недостаточное количество тромбоцитов (тромбоцитопении);
б) функциональная неполноценность тромбоцитов (тромбоцитопатии);
в) сочетание количественной и качественной патологии тромбоцитов.
III. Геморрагические диатезы, проявляющиеся в результате избыточного фпбринолиза:
а) эндогенного;
б) экзогенного.
IV. Геморрагические диатезы, проявляющиеся в результате патологии сосудистой стенки:
а) врожденные;
б) приобретенные.
V. Геморрагические диатезы, развивающиеся в результате сочетания нескольких причин (тромбогеморрагический синдром, болезнь Виллебранда).
Наиболее частой причиной геморрагических диатезов является дефект тромбоцитарного звена гемостаза, который является причиной кровоточивости у 80% больных [Марквардт Ф., 1976]. В группе больных геморрагическими диатезами, развивающимися при неполноценности прокоагулянтного звена гемостаза, наиболее часто диагностируется гемофилия А (65—80%), гемофилия В (13—18%) и гемофилия С (1,4—9%).
Исторически же сложилось так, что большую известность приобрели геморрагические диатезы, обусловленные дефектом формирования фибрина. В настоящее время известно, что образование фибрина обеспечивается путем правильного взаимодействия белков прокоагулянтов, большинство из которых имеет собственный номер, указываемый римской цифрой. Различают 13 субстанций, среди которых фибриноген (фактор I), протромбин (II), проакцелерин-акцелерин (V), проконвертин (VII), антигемофилический глобулин А (VIII), кристмас-фактор (IX), фактор Стюарта — Прауэра (X), предшественник тромбопластина плазмы (XI), фактор Хагемана (XII), фибринстабилизирующий фактор (XIII). Кроме них, три обнаруженных за последнее время фактора не имеют цифрового обозначения. Это факторы Флетчера, Фицжеральца и Пассовой.
Количественный или качественный дефект любого из указанных выше прокоагулянтов, а также появление в крови больного ингибитора этого фактора способны вызвать геморрагическое состояние у больного.
Большое количество этих состояний, которое подходит к цифре 30, а также большое сходство их клинических проявлений позволяют объединять данные заболевания под общим названием «гемофилии».
Для гемофилий характерны обширные, глубокие, обычно изолированные, спонтанные кровоподтеки и гематомы, частые кровоизлияния в суставы при крайне редком развитии кожных и слизистых «пурпур» в редких и слабо выраженных кровотечениях при поверхностных повреждениях кожи. Грубые лабораторные пробы демонстрируют удлинение времени свертывания крови при отсутствии нарушения времени кровотечения. Практические врачи должны четко представлять, что точная диагностика причины геморрагического диатеза возможна лишь при применении специальных лабораторных методов исследования, без чего адекватная терапия практически невозможна.
Среди геморрагических диатезов, развивающихся при неполноценности тромбоцитарного звена гемостаза, наиболее часто встречаются те, которые обусловлены уменьшением количества тромбоцитов в кровотоке больного. Эти состояния, называемые синдромом Верльгофа, неоднородны по своей причине. Количество тромбоцитов может снижаться как в результате образования против них аутоантител (аутоиммунные тромбоцитопении), так и в результате неполноценного образования их в костном мозге. Возможны также неполноценность тромбоцитарной оболочки и их цитолиз.
Внимание клиницистов за последние годы оказалось прикованным к таким геморрагическим состояниям; которые обусловлены функциональной неполноценностью тромбоцитов, не способных обеспечивать полноценный гемостаз даже при достаточном их количестве в кровотоке больного. После того как такая патология впервые была описана Глянцманом, обнаружено большое количество патологических форм, которые обусловлены нарушением того или иного этапа формирования тромбоцитарной пробки, осуществляемого тромбоцитами: их адгезией, агрегацией, активацией прокоагулянтного звена, ретракцией кровяного сгустка.
Обнаружение этих дефектов, выявление их сочетаний с некоторыми другими проявлениями заболевания привело к описанию ряда отдельных нозологических форм. В то же время изучение функций тромбоцитов при ряде описанных заболеваний позволило отметить отсутствие связи между нарушениями тромбоцитарных функций и другими симптомами, не имеющими отношения к гемостазу.
Различные сочетания дефектов тромбоцитарных функций позволили говорить о наличии целой группы тромбоцитопатий, проявляющихся самыми разнообразными соединениями нарушений таких функций тромбоцитов, как адгезия, агрегация, реакция освобождения, активация прокоагулянтов, ретракция. При уточнении причины геморрагического диатеза необходимо детальное изучение как количественного, так и качественного состояния тромбоцитов в условиях лаборатории.
В клинической картине этих заболеваний характерными являются частые длительные кровотечения при поверхностных повреждениях кожи, частые кожные и слизистые «пурпуры», в то время как кровоизлияния в суставы, спонтанные кровоподтеки и гематомы довольно редки.
Дефекты гемостаза, вызванные патологией сосудистой стенки, диагностируются довольно легко в тех случаях, когда эта патология доступна визуальному наблюдению: при болезни Рандю— Ослера, синдроме Элерса—Данлоса, болезни Гиппеля—Линдау, синдроме Казабаха—Мерритта и др. В настоящее время имеются указания, что геморрагические диатезы могут развиваться при неполноценности коллагена сосудистой стенки и нарушенной вследствие этого адгезии тромбоцитов. Однако эту патологию возможно диагностировать только при применении сложных лабораторных методов.
Большое внимание клиницистов за последнее время привлекают случаи развития геморрагий у больных при множественном микротромбировании капилляров внутренних органов. Эти состояния получили названия тромбогеморрагического синдрома. Патогенез его объясняется тем, что при массивном быстром тромбообразовании в сгустке потребляются многие факторы свертывания крови, особенно тромбоциты и фибриноген. Кроме того, гипоксия сосудистой стенки приводит к выбросу в кровоток большого количества активаторов плазминогена и повышению фибринолитической активности крови. Диагностика этих состояний весьма важна, так как требует «парадоксального» применения антикоагулянтов для лечения геморрагий.
Интересные находки обнаружены при изучении патогенеза кровоточивости у пациентов с болезнью Виллебрандта, которая характеризуется сочетанием симптомов, отражающих нарушения как прокоагулянтного, так и тромбоцитарного звеньев гемостаза. При этом было выявлено, что антиген фактора VIII имеет существенное значение для запускания адгезии тромбоцитов к поврежденной поверхности и показал важность взаимоотношений этих ведущих механизмов остановки кровотечений.
Большое многообразие причин геморрагических диатезов, создание специфических методов лечения этих состояний обязывают практических врачей к детальному изучению вопросов диагностики и лечения больных с повышенной кровоточивостью.
Наиболее частыми глазными проявлениями при пурпуре являются подкожные и конъюнктивальные геморрагии. Кровоизлияния в сетчатку весьма редки. В тех случаях, когда они все же есть, геморрагии располагаются в слое нервных волокон. Следует иметь в виду, что при травме глаза, в том числе и хирургической, возможно обильное кровотечение, особенно при гемофилии.
---
Статья из книги: Патология органа зрения при общих заболеваниях | Комаров Ф.И., Нестеров А.П., Марголис М.Г., Бровкина А.Ф.

Комментариев 0