Некоторые синдромы общих заболеваний глаз, уха, полости рта

Описание

К их числу прежде всего относятся дефекты развития и заболевания, связанные с генетическими аномалиями семейно-наследственного характера.
Синдром Lobstein (1895), Van-der-Hoeve, de-Klein (1918). Это системное семейно-наследственное заболевание, связанное с нарушением развития мезодермы. Его признаки: наиболее часто наблюдаются синие склеры, затем ломкость костей и реже всего тугоухость. Так, Dessoff лишь у 60% пациентов с синими склерами наблюдал нарушение слуха, только когда они достигли 20-летнего возраста. По Van-der-Hoeve, три степени тугоухости проявлялись у больных в возрасте 14, 20 и 30 лет.
Тугоухость имеет характер отосклероза, частично со вторичным заболеванием лабиринта. Описана сине-серая окраска барабанной перепонки (Cornil, Berthier, Sild, Funk, no Schroder), соответствующая окраске склеры, в обоих случаях из-за ненормальной тонкости тканей. Fowler обследовал 548 пациентов на отосклероз и обнаружил его у 3% лиц с белой склерой, у 12%— при неясно синей, у 50%—с явно синей, у 100%—с фиолетовой. Преимущественно болеют женщины, особенно начиная с периода полового созревания. Возможна комбинация голубых склер с пигментной дистрофией сетчатки.
Мраморная болезнь костей (болезнь Альберс-Шенберга). Заболевание связывают с дисгармонией в развитии костной и кроветворной ткани в связи с нарушением энхондрального окостенения под влиянием наследственных факторов.
Основные симптомы болезни: ломкость костей, анемия без существенных изменений лейкопоэза, массивное накопление костной субстанции, резкая плотность, гомогенность, бесструктурность костей скелета, напоминающая мрамор, отсюда и название болезни. Возможен кариозный процесс зубов, осложненный остеомиелитом челюстей и некрозом. Pietruschka (1953) наблюдал у больных понижение слуха в связи с поражением внутреннего уха.
Офтальмологические симптомы проявляются экзофтальмом, вследствие уплощения глазницы и изменения костей черепа, расходящимся косоглазием, застойными сосками и впоследствии атрофией зрительного нерва. Л. А. Дымшиц с сотрудниками (1962), кроме того, наблюдал у ребенка тапеторетинальную дегенерацию типа punctata albescens.
Нарушения в развитии кроветворной системы, глаз, ушей и полости рта описаны в следующем синдроме.
Синдром Chaufford—Minkowski (1900)— Ganssein (1922) — конституциональная гемолитическая анемия, желтуха, спленомегалия и ряд врожденных расстройств: отосклероз, микрофтальм, врожденная катаракта, гетерохромия радужки, цветовая слепота, аномалии зубов и ушей, синдактилия, полидактилия.
Синдром Francescetti— Kleina (1949) — заболевание, для которого типично двустороннее, а реже одностороннее неполное или абортивное неправильное образование наружного, а иногда и среднего уха, тугоухость в сочетании с поражением век и disostosis mandibulo facialis. У больных бывает монголоидныи разрез глазной щели, возможна колобома латеральной части нижнего, реже верхнего века, птоз, парез глазодвигательных мышц, гипоплазия костей лица, преимущественно нижней челюсти и скуловой кости, высокое небо (макростомия), плохое смыкание челюстей, слепые фистулы между углами рта и ушами, возможны и другие деформации лицевого скелета, ненормальный рост волос на лице с языкообразным направлением в сторону щек.
Умственное развитие замедлено, что иногда можно связать с расстройством слуха.
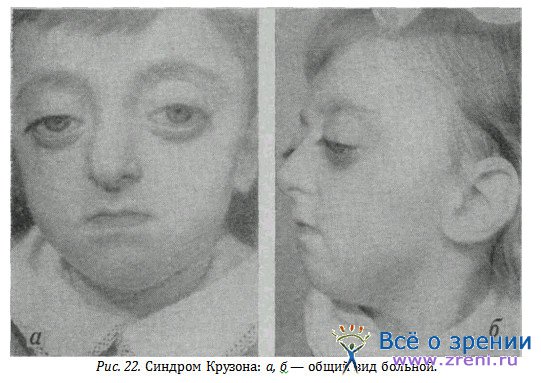
Черепно-лицевой дизостоз (болезнь Крузона) описан как самостоятельный синдром. Заболевание возникает вследствие неправильного развития костей черепа и лица и имеет следующие симптомы: башенный или круглый череп, лоб, выдающийся вперед и суживающийся к средней линии, аплазию и западение кзади верхней челюсти, расширение корня носа и прогнатизм нижней челюсти, поэтому лицо в профиль похоже на клюв птицы («попугайная болезнь»), глазницы плоские, экзофтальм, иногда вывих глазных яблок из орбит, расходящееся косоглазие. Вначале бывает застойный сосок, а затем вторичная и первичная атрофия зрительного нерва и соответствующие нарушения остроты зрения и поля зрения. Неврологические симптомы проявляются признаками гидроцефалии, эпилепсии, возможного умственного недоразвития.
У одной больной, лично наблюдаемой, симптомы заболевания не были резко выражены. Девочка не проявляла признаков умственного недоразвития. Главными симптомами были изменение лицевого скелета, экзофтальм (рис. 22 а, б), застойные соски на фоне гидроцефального синдрома. После двусторонней декомпрессивной трепанации черепа, последний, а также и застойные соски регрессировали и уже длительно стабилизировалась острота зрения в 0,7 и 1,0.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
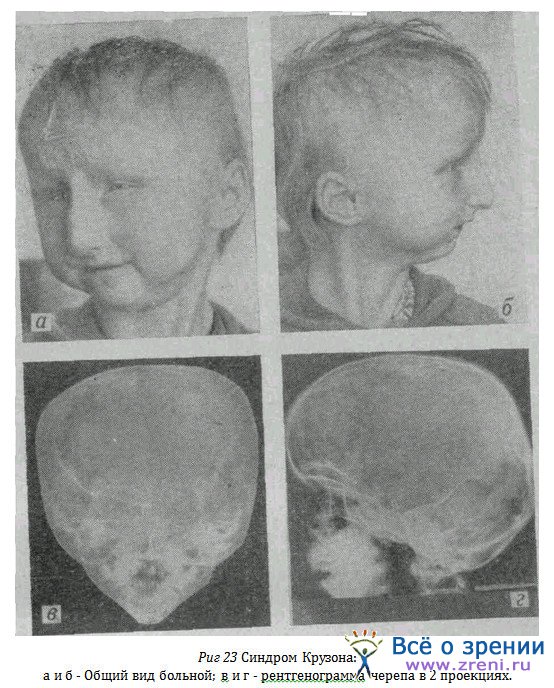
У другой больной—резко выраженный синдром (рис. 23 а, б) с типичными изменениями лицевого скелета, всего черепа (рис. 23в, г), с выраженным недоразвитием и глухотой. Острота зрения равна 0,01, вследствие еще врожденной катаракты.
Пигментная дистрофия сетчатки. Еще Graefe (1895) указал на сочетание пигментной дистрофии и глухонемоты. Типичное поражение сетчатки как проявление наследственного признака, передающегося по рецессивному признаку, часто сочетается с тугоухостью, глухотой или глухонемотой, возможной дебильностью или идиотией, полидактилией и другими изменениями скелета.
Kjerrumgaard из числа больных с пигментной дистрофией нашел у 28% расстройство слуха, у 17% — неврологические и у 12%—эндокринологические расстройства. Hallgren наблюдал синдром пигментной дистрофии и врожденной глухонемоты у 177 членов 102 семейств с семейным сходством степени тугоухости. У 87% этих пациентов авторы обнаружили вестибулярно-мозжечковую атаксию и полную глухоту—у 12,4%.
Francescetti и Klein (1947, по Е. И. Стародубцевой) наблюдали семью, в которой от брака мужчины, происходящего из семьи с пигментной рецессивной ретинопатией, и женщины с наличием доминантного наследственного отосклероза родилось 8 детей, из которых у 5 была пигментная дистрофия с отосклерозом. Эти же авторы (по Schroder) обнаружили у отца и сына с такими заболеваниями эквивалентные поражения одного гена.
У обитателей домов глухонемых пигментная дистрофия была обнаружена от 2,5% (Hocquard, Mulder) до 10% (Lemke, no Schroder) и вдвое чаще у лиц с врожденной глухонемотой. У последних пигментная дистрофия встречается в 1000 раз чаще, чем у людей с нормальным слухом (Perrin).
У глухонемых возможны атипичные и стертые формы пигментной дистрофии, а также центральные хориоретиниты (Cornea, Teodorescu, Banee) вне связи степени глухоты с тяжестью хориоретинального процесса. Bunge у глухого больного, страдающего наследственной нефропатией, обнаружил degeneratio retinae punctata albescens (синдром Alport).
У наблюдаемых лично брата и сестры, воспитанников школы глухонемых, страдающих пигментной дистрофией, были положительными аллергическая и серологические реакции на токсоплазмоз.
Пигментная дистрофия сетчатки может сочетаться с другими синдромами.
Синдром Laurence (1866)—Moon (1866)— Bardet (1920) — Вidle (1922) — наследственное заболевание, передается по рецессивному типу и обусловлено множественными дефектами гена.
Заболевание связано с абиотрофией энцефалогипофизарной области и ретинального нейроэпителия. Оно имеет следующие признаки: нарушение жирового обмена, половые расстройства (distrophia adiposogenitalis), глухоту, пигментную дистрофию сетчатки, могут быть и другие дефекты развития глаз (косоглазие, колобома желтого пятна, катаракта, нистагм, микрофтальм и др.), врожденные пороки сердца, деформацию черепа, низкий рост, умственные расстройства и др.
Начинается в раннем детском возрасте, но основные проявления чаще возникают между 10 и 15 годами жизни.
Лично пришлось наблюдать больного, у которого этот синдром в стертом виде проявился лишь в 25-летнем возрасте (ожирение, пигментная дистрофия).
Синдром Ушера — врожденная глухота и пигментная дистрофия сетчатки.
Синдром Гурлера — липодистрофия вследствие расстройства жирового обмена. У этих больных карликовый рост и неправильная пропорциональность отдельных частей тела, гепато- и спленомегалия, отставание в интеллектуальном развитии, врожденная глухота, мегалокорнеа и помутнение роговиц.
Нейрофиброматоз — болезнь Реклингаузена (1882).
Это семейно-наследственное заболевание, проявляется уже у новорожденных детей или несколько позже. Характерно развитие множественных опухолевидных фиброзных разрастаний из оболочек черепно-мозговых, спинномозговых, периферических и симпатических нервов, пигментные пятна кожи цвета «кофе с молоком», остеодистрофия в разных отделах черепа, особенно лицевого скелета, асимметричные дефекты лобной, затылочной, височной костей, глазницы. В последнем случае или при опухолевидном разрастании цилиарных нервов возникает экзофтальм. Нейрофибромы могут быть в веках, конъюнктиве, склере, радужке, в сетчатке, на соске, хиазме.
Возможно развитие нейрофибромы слухового нерва с симптомами внутричерепной опухоли, в том числе парезом отводящего нерва, паралитическим косоглазием, односторонней глухотой, вестибулярными симптомами. Повышение внутричерепного давления проявляется головокружением, головной болью, шаткой походкой, рвотой, снижением остроты зрения и застойным соском. На рентгенограмме обнаруживается расширение внутренних слуховых проходов, деструкция вершины пирамиды височной кости, а у некоторых — расширение канала зрительного нерва, верхней орбитальной щели.


Бывают нарушения психики и эпилептиформные припадки.
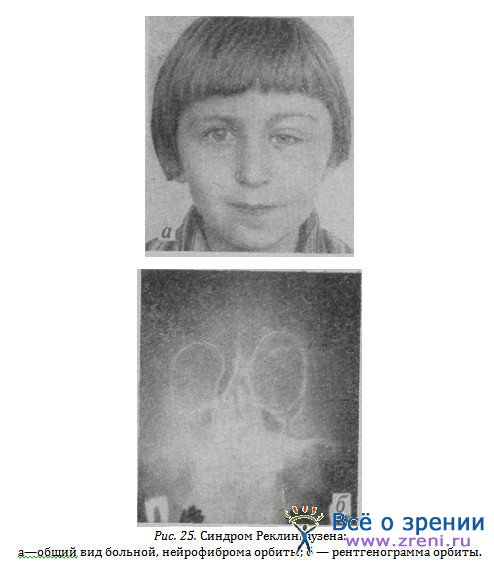
Иллюстрации собственных наблюдений у одной больной показывают типичные изменения кожи (рис. 24, а} в сочетании с нейрофибромой век (рис. 24, 6} и конъюнктивы (рис. 24, в). У другой была нейрофиброма век (рис. 25, а), орбиты (рис. 25, б), основания черепа без изменений глаза; у третьей больной—опухоль орбиты с быстрым ростом, симулирующая злокачественную (рис. 26). Слепой глаз был удален. Гистологически установлен диагноз: нейрофиброма зрительного нерва.
Триада Hutchinsona — бочкообразная форма и полулунные дефекты режущего края верхних центральных резцов, реже — боковых верхних или центральных нижних, паренхиматозный кератит и глухота— проявления врожденного сифилиса.
Ряд синдромов или заболеваний глаз и уха может быть приобретенным в зависимости от инфекционных, токсических влияний, а также от не установленных еще причин.
Синдром Hogana (1945) — токсическое или инфекционное повреждение периферических рецепторов кохлеарного и вестибулярного аппаратов, вследствие чего возникает шум в ушах, быстро прогрессирующая потеря слуха, головокружение, нарушение равновесия, аудиометрически определяется ретрокохлеарное расстройство восприятия. Офтальмологические симптомы: двусторонний интерстициальный кератит, для которого характерна глубокая инфильтрация роговицы различной интенсивности в отдельных участках, поздняя глубокая васкуляризация. Реакция Вассермана — отрицательная. Иногда синдром бывает связан с узелковым периартериитом. Автор считает описанный им синдром проявлением системного кардиоваскулярного заболевания, отличающегося по течению от узелкового периартериита. Прогноз для жизни и зрения хороший, плохой — для слуха.
Лабиринтным расстройствам (Mayoux, 1953, по Темпля) у больных, страдающих головокружением без общих сосудистых причин, сопутствуют изменения давления во внутренней слуховой и центральной артерии сетчатки. Последнее проявляется сужением и ригидностью артерий сетчатки, иногда легким отеком ее ткани, расширением вен или стазом крови в них, повышением или понижением ретинального давления. Ввиду однотипного кровоснабжения лабиринта и сетчатки по состоянию ее сосудов можно судить о сосудах лабиринта.
Синдром Фогта—Койанаги (1914), синдром Харада (1926). В настоящее время оба синдрома рассматривают как варианты одного заболевания — увеоэнцефалита инфекционно-аллергической природы, а Hager (1957) причисляет к ним и симпатическое воспаление. Основным отличием обоих синдромов от симпатического воспаления является отсутствие предшествующей травмы глаза. Впервые М. Л. Краснов (1935) описал типичный синдром Фогта — Койанаги, возникший у больного спустя много лет после проникающей травмы глаза.
Ведущими симптомами синдромов Фогта — Койанаги и Харада является увеит и менингоэнцефалит. Заболевают чаще мужчины среднего возраста. Начинается болезнь остро, общим недомоганием, головной болью, повышением температуры, рвотой и неврологическими признаками базального серозного менингита или менингоэнцефалита. Зрение при этом понижено. При офтальмоскопии отмечается отек зрительного нерва.
На 2—4-й неделе остро развивается увеит, в случае возможности офтальмоскопии обнаруживается картина эксудативного хориоидита, невроретинита, вплоть до отслойки сетчатки (Харада), склонной к самопроизвольному прилеганию. Зрение резко снижается или больные слепнут вследствие осложнения переднего увеита (заращение зрачка, осложненная катаракта, вторичная глаукома) или атрофии зрительных нервов. Спустя несколько недель после начала заболевания возникают трофические изменения кожи и волос: поседение волос, бровей и ресниц, лейкодермия, гнездное выпадение волос на голове. Более чем у половины больных возникает головокружение, шум в ушах, ухудшение слуха, вследствие поражения среднего уха или основного ствола слухового нерва. Противоречивы мнения авторов о возможности восстановления слуха.
И. А. Завьялов (1969) описал 5 больных с этим синдромом, однако ни у одного больного в описании истории болезни не указано понижение слуха.
Алкогольный кохлеарный неврит нередко развивается одновременно с токсическим невритом зрительного нерва. Глухота может сочетаться с глазодвигательными расстройствами.
Интоксикация свинцом может быть причиной неврита слухового и зрительного нервов. Одновременно и внезапно снижается острота зрения и слух, появляется шум в ушах, расстройство равновесия, зрительные и слуховые галлюцинации, которые могут стать постоянными, если интоксикация продолжается.
У лиц с повышенной чувствительностью к стрептомицину или с применением его больших доз может наступить атрофия зрительных и слуховых нервов.
При некоторых заболеваниях одновременно или последовательно вовлекаются в общий процесс (чаще инфекционный) глаза и ротовая полость.
Синдром Стивенса — Джонсона — эрозивный эктодермоз, дерматостоматит (1922). Заболевание развивается на фоне аллергии, провоцируется инфекциями или медикаментами, действующими как аллергены, имеет острое начало, с высокой температурой, головной болью, ангиной, ревматоидными симптомами. Характерна полиморфная сыпь в виде рассеянных, резко ограниченных насыщенно красных папул, пузырей, язв, преимущественно на коже лица, разгибательной поверхности рук и ног, тыла кистей и стоп. На конъюнктиве и слизистой рта, носа, половых органов образуются пузыри, а затем и язвы, что вызывает слипание обнаженных поверхностей. Возможно поражение роговицы, на которой возникают эрозии, изъязвление, вплоть до образования в исходе бельм роговицы.
В наблюдении А. Г. Маркзицера описано у больного поражение слизистой щек, носоглотки, миндалин, задней стенки глотки.
Тяжелые и распространенные поражения привелось наблюдать и в личной практике у 2 больных ангиной, после приема тетрациклина.
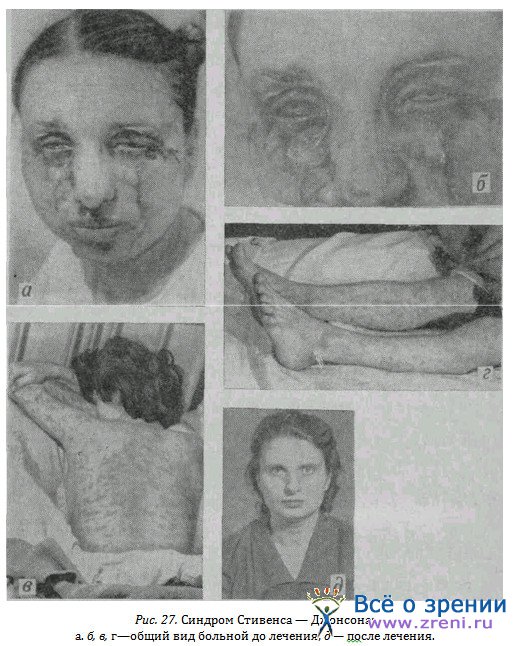
Больная Т., 32 лет, медсестра, заболела ангиной, на протяжении дня приняла 3 таблетки тетрациклина. На 2-й день наступили все описанные выше симптомы (рис. 27 а, б, в, г), появилась сыпь по всему телу, пузыри на слизистой век, обоих глаз, губ, десен, щек, зева, глотки, миндалин, половых органов, прямой кишки. Спустя 2 дня наступило изъязвление роговицы левого глаза. Излечение (рис. 27, д) наступило в инфекционной клинике под влиянием массивной симптоматической терапии и, кроме того, больших доз кортикостероидов, витаминов и др.
Больной М., 8 лет, за 3 месяца до настоящего заболевания перенес ангину, участковый врач назначил прием тетрациклина. После 1 таблетки появилась кожная сыпь, и препарат был отменен. Ребенок выздоровел. При повторном заболевании ангиной тот же врач вновь назначил прием тетрациклина. Прием 1 таблетки привел к появлению кожной сыпи, уже с поражением слизистых.
При осмотре ребенка в детской клинике через неделю после начала заболевания обнаружилась гиперемия и дефекты слизистой век, мягкие спайки с конъюнктивой глаза, несмотря на частое их разделение. Состояние слизистой рта — то же, что и у предыдущей больной. Излечение наступило только после включения в арсенал средств кортикостероидов.
В обоих случаях, очевидно, имела значение лекарственная аллергия. В. И. Кобзева (1968) наблюдала 3 больных с этим синдромом, также вследствие лекарственной аллергии. Из них у 1 больного такое состояние возникло после применения норсульфазола, у 1 — салицилатов и у 1 — пенициллина и реопирина.
Болезнь Бехчета — заболевание вирусной этиологии, характерно рецидивирующим течением и обилием различных симптомов, но основные—это афтозный стоматит, язвенный процесс в области наружных половых органов, увеит с гипопионом, часто заканчивающийся слепотой. Позже присоединяются поражения суставов, вен, тонзиллит. Особенно серьезны изменения центральной нервной системы (менингоэнцефалит), которые бывают причиной смертельных исходов (Evans с соавторами, 1957). Чаще болеют мужчины, преимущественно молодого возраста.
Необязательно появление сразу всей триады симптомов, они могут появляться разновременно. Кроме того, к настоящему времени накопились сведения о значительно большем разнообразии глазных симптомов, таких, как мембранозный конъюнктивит с изъязвлением конъюнктивы и образованием спаек (М. М. Золотарева, Н. Ф. Павлов, М. С. Завадская, Л. Ф. Пильтиенко, 1960; 3. К. Бекназарова, Р. Л. Григорьянц и Э. Н. Черняк, 1961); поражение заднего отрезка глаза в виде эксудативного хориоретинита, нейроретинита, перифлебита с кровоизлияниями в сетчатку и стекловидное тело, дегенерация сетчатки и др. Эти изменения могут предшествовать клиническим симптомам переднего увеита, но могут остаться нераспознанными вследствие помутнения стекловидного тела. Доказательством этому служат клинические наблюдения и выделение вируса из стекловидного тела и субретинальной жидкости. Описаны различные течения заболевания.
В личном наблюдении у 1 больного кожа была в норме, минимальные нарушения наблюдались в слизистой рта. Изменения глаз отличались своей тяжестью и проявлялись комбинацией тяжелого рецидивирующего гипопионувеита и центрального хориоретинита, вторичной глаукомой. На протяжении 2 лет острота зрения снизилась до 0,01—0,02. Наряду с этим у больного появились явные нарушения психики, по поводу чего он несколько месяцев лечился в психиатрической больнице.
А. И. Лопотко (1966) описал больного, у которого не было изменений глаза и на фоне лишь умеренного поражения кожи плеч и бедер возникли такие проявления трахеобронхита, что больному была дважды сделана трахеотомия.
Синдром Съегрена — сухой кератоконъюнктивит. Для заболевания характерны прогрессирующая атрофия и недостаточность функции желез: слезных, слюнных, верхних дыхательных и пищеварительных путей. Заболевание чаще развивается у пожилых женщин в периоде климакса, на фоне ревматоидного артрита и анемии, имеет хроническое течение. Описаны заболевания и у молодых женщин без эндокринных нарушений. Ныне этот синдром связывают с коллагенозом, которому способствуют нарушения функции печени, витаминного обмена, эндокринная недостаточность, инфекции.
У больных возникает сухость во рту, носу, зеве, носоглотке, понижение слюноотделения и разрушение зубов, часто опухание околоушных узлов, сухость волос, кожи, ломкость ногтей и др.
Особенно типичны жалобы больных на сухость и зуд в глазах, светобоязнь, ощущение инородного тела и появление тягучего сероватого нитчатого секрета в конъюнктиве. Это спущенный эпителий и слизь с конъюнктивы и роговицы. При потягивании нитей они оказываются эластичными и при их опускании возвращаются в исходное состояние. После удаления нитей остаются эрозии конъюнктивы и роговицы, а в дальнейшем развивается ее помутнение.
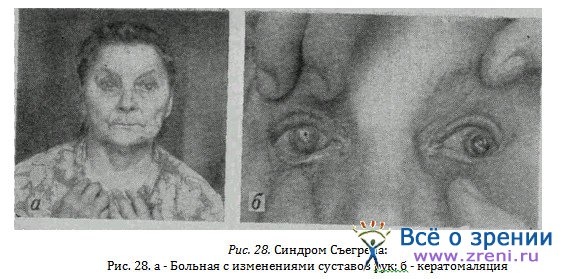
Личные наблюдения показывают возможность стертого синдрома, когда у больных есть лишь сухость и изменения конъюнктивы и, напротив, очень тяжелого симптомокомплекса.
У больной Б. 65 лет, страдающей типичным ревматоидным артритом с поражением мелких суставов (рис. 28. а) слизистой глаз, рта, пищеварительных и дыхательных путей, возникла кератомаляция обоих глаз (рис. 28, б).
Энцефалотригеминальный ангиоматоз (болезнь Стюржа—Вебера). Этот синдром проявляется ангиомой половины лица в зоне иннервации тройничного нерва с вовлечением кожи и слизистой век и часто односторонней глаукомой соответственно этой половине лица. Кроме того, обычно у больного имеется ангиома оболочек и вещества головного мозга очаги обызвествления в нем. Поэтому у больных наблюдается очаговая симптоматика, соответственная локализации ангиомы. Возможны эндокринные расстройства в том числе ожирение. В литературе не встретилось описания при этом синдроме поражения ЛОР-органов. Поэтому большой интерес представляет личное наблюдение.
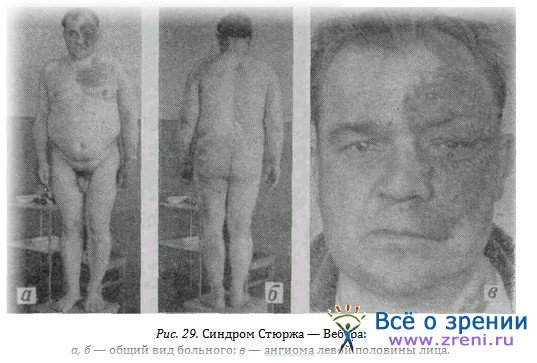
Больной Г., 48 лет, поступил в ЛОР- клинику для хирургического вмешательства с диагнозом полипоз носа. До этого у больного внезапно появилось сильное кровотечение из левой ноздри, которое с трудом удалось остановить, и после этого исчезли «полипы». Ввиду жалоб больного на пониженное зрение и видимые изменения кожи лица он был направлен в глазную клинику на консультацию.
Обнаружена (рис. 29, а) ангиома левой половины лица, включая нос, кожу и слизистую век, глазного яблока, кожу туловища, ожирение (рис. 29 б, б). Изменений центральной нервной системы и глаукомы не выявлено. Снижение остроты зрения связано с аномалией рефракции. Ангиома слизистой носа была ошибочно принята за полип.
---
Статья из книги: Офтальмологические симптомы при болезнях ЛОР-органов и полости рта | Золотарева М.М.

Комментариев 0