Редкие формы глаукомы


Содержание:
Описание
Среди всех видов глаукомы выделяют группу так называемых редких форм, составляющих 2,0~2,5% от данной нозологии. Их социальное и медицинское значение особенно велико в связи с тем, что формирование глаукомы происходит в детском и юношеском возрасте. Единой общепринятой классификации этих заболеваний в настоящее время нет. По механизму развития редкие формы глаукомы подразделяют, согласно рекомендациям Европейского общества глаукоматологов (EGS, 2003), на следующие группы.- Глаукома, связанная с мезенхимальным дисгенезом - синдром Аксенфельда, синдром Ригера, синдром Петерса, синдром Франк-Каменецкого.
- Вторичная закрытоугольная глаукома с прогрессирующим формированием эндотелиальной мембраны — иридокорнеальный эндотелиальный синдром (синдром Чандлера, синдром Когана-Риза, прогрессирующая эссенциальная мезодермальная атрофия радужки).
- Вторичная открытоугольная глаукома, офтальмологически обусловленная синдром пигментной дисперсии.
↑ ГЛАУКОМА, СВЯЗАННАЯ С МЕЗЕНХИМАЛЬНЫМ ДИСГЕНЕЗОМ
Мезенхимальный дисгенез может поражать лишь УПК или передний сегмент полностью. Упрощённо спектр патологий мезехимального дисгенеза распределён по лестничной схеме классификации Waring (рис. 33-48).
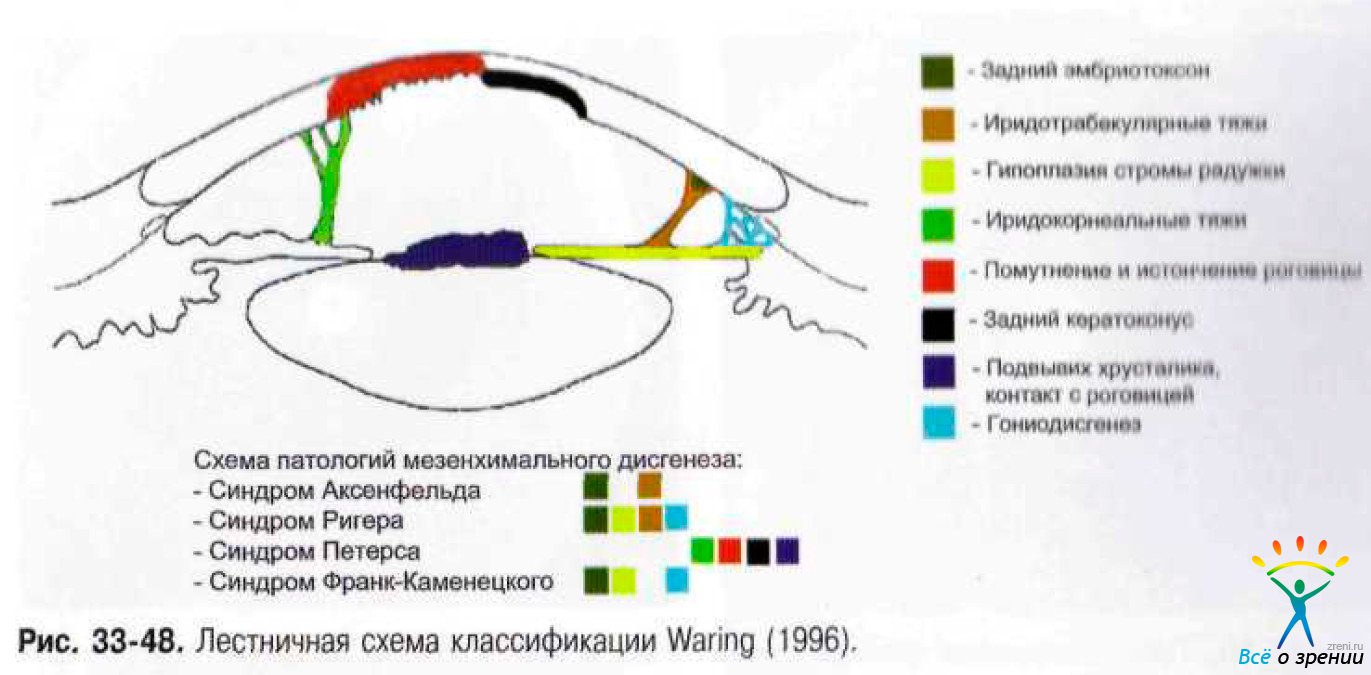
↑ Синдром Аксенфельда-Ригера
Дисгенез Аксенфельда — состояние, при котором выявляют аномальные тяжи радужки, проходящие через УПК и прикреплённые к выступающей в переднюю камеру линии Швальбе (задний эмбриотоксон). Если этому сопутствует глаукома, аномалию называют синдромом Аксенфельда (рис. 33-49).

Синдром Ригера состояние, при котором обнаруживают офтальмольмоскопические изменения, типичные для синдрома Аксенфельда, в сочетании со скелетными аномалиями (например, челюстная гипоплазия, микродентизм) и другими пороками развития.
↑ КОД ПО МКБ 10
Н40.5 Глаукома вторичная вследствие других болезней глаз
Н21.2 Дегенерация радужной оболочки и цилиарного тела.
Н21.5 Другие виды спаек и разрывов радужной оболочки и цилиарного тела.
↑ ЭПИДЕМИОЛОГИЯ
Наблюдают одинаково часто у мужчин и женщин. Может иметь спорадическое происхождение или наследоваться по аутосомно-доминантному типу. В 60% случаев развивается глаукома.
↑ ЭТИОЛОГИЯ
Выявлена патология хромосомы 6, делеция хромосомы 13, в 30% случаев диагностируют вновь возникшие мутации и спорадическое появление синдрома.
↑ ПАТОГЕНЕЗ
Недоразвитие дренажной системы, а также сращение радужки с кольцом Швальбе приводит к снижению оттока внутриглазной жидкости и развитию глаукомы.
↑ КЛИНИЧЕСКАЯ КАРТИНА
Для синдрома Ригера характерны гипоплазия верхней челюсти, широкая переносица, короткий губной желобок, а также патология зубов — маленькие конической формы зубы с широкими промежутками в зубном ряду (рис. 33-50) и частичная адентия. У больных выявляют пупочную и паховую грыжи, гипоспадию, гормональную недостаточность, пороки клапанов сердца.

↑ ДИАГНОСТИКА
Диагностика основана на данных соматического и офтальмологического обследования.
↑ Анамнез
Выявляют отягощённую по глаукоме наследственность и жалобы на низкое зрение с детства.
↑ Инструментальные исследования
При офтальмологическом обследовании выявляют:
- миопическую рефракцию высокой степени;
- при биомикроскопии: гипоплазию мезодермального листка радужки, задний эмбриотоксон и иридотрабскулярные тяжи к линии Швальбе, в некоторых случаях - помутнение роговицы на периферии, задний кератоконус, врождённую катаракту (иногда в сочетании с колобомой радужки);
- при офтальмоскопии: колобому сосудистой оболочки, косой врез и миелиновые волокна ДЗН, отслойку сетчатки;
- при гониоскопии: частично или полностью закрытый иридотрабекулярными тяжами угол, выдающуюся в переднюю камеру линию Швальбе, уплотнение трабекулы.
Ультразвуковая биомикроскопия позволяет выявить иридотрабскулярные тяжи в УПК (рис. 33-51).

↑ Дифференциальная диагностика
Проводят с синдромом Франк-Каменецкого.
↑ Показания к консультации других специалистов
Необходима консультация травматолога-ортопеда, стоматолога, кардиолога, эндокринолога, хирурга, педиатра.
↑ Пример формулировки диагноза
Врождённая декомпенсированная глаукома, синдром Аксенфельда.
↑ ЛЕЧЕНИЕ
При развитии вторичной глаукомы показана фистулизирующая антиглаукомная операция, как дополнение - медикаментозная гипотензивная терапии.
↑ Дальнейшее ведение
При необходимости пожизненная медикаментозная гипотензивная терапии или хирургическое лечение глаукомы и сопутствующей глазной патологии, терапия, направленная на улучшение трофики, 2 раза в год.
↑ ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Необходимость диспансерного наблюдения не реже одного раза в 3 мес с регулярным контролем зрительных функций и ВГД.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
↑ ПРОГНОЗ
При синдроме Аксенфельда снижение зрительных функций может происходить при декомпенсации глаукомного процесса.
При синдроме Ригера врождённые низкие функции обусловлены сочетанной глазной патологией. Прогноз для зрения неблагоприятный, так как данную форму глаукомы считают рефрактерной.
↑ Аномалия Петерса
Аномалия Петерса - состояние, при котором выявляют врождённое помутнение центральной части роговицы в сочетании с мезенхимальным дисгенезом радужки и эктопией хрусталика.
↑ КОД ПО МКБ-10
Н40.5 Глаукома вторичная вследствие других болезней глаз.
H21.2 Дегенерация радужной оболочки и цилиарного тела.
Н21.5 Другие виды спаек и разрывов радужной оболочки и цилиарного тела.
Q13.3 Врождённое помутнение роговицы.
Q13.4 Другие пороки развития роговицы.
↑ ЭПИДЕМИОЛОГИЯ
Большинство случаев аномалии Петерса считают спорадическими, хотя описаны рецессивный и иррегулярный доминирующий виды наследования. Примерно 80% описанных случаев двусторонние.
↑ ЭТИОЛОГИЯ
Одна из теорий возникновения дисгенеза Петерса - прекращение нормальной функции развивающегося эндотелия в сочетании с внутриутробным подвывихом хрусталика, произошедшим до или после его полного развития.
↑ ПАТОГЕНЕЗ
Грубая врождённая деформация УПК приводит к формированию плоскости иридокорнеальных сращений и нарушению опока внутриглазной жидкости.
↑ КЛИНИЧЕСКАЯ КАРТИНА
Характерны низкий рост, расщелины губы или нёба, заболевания органа слуха и задержка умственного развития. Различают два клинических варианта заболевания.
- Синдром Петерса типа I - типичное нубекулярное (облачковидное) центральное помутнение роговицы, окаймлённое тяжами радужки (рис. 33-52), которые пересекают переднюю камеру от зрачкового пояса радужки до роговицы. Хрусталик прозрачный с правильным расположением. Глаукома присоединяется в 30% случаев.
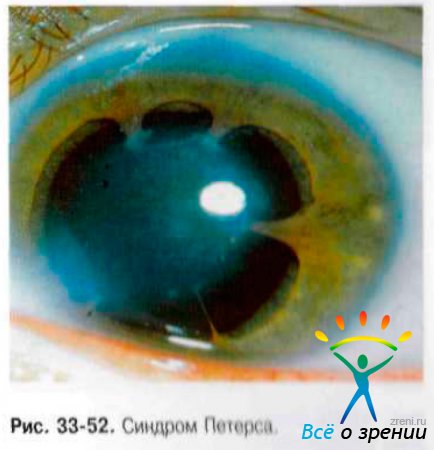
- Синдром Петерса типа II - тип тяжёлого течения заболевания, при котором происходит сращение хрусталика с центральной корнеальной лейкомой и формирование передней полярной катаракты. Часто ассоциирован с микрокорнеа, микрофтальмом. плоской роговицей, склерокорнеа, колобомой, аниридией и дисгенезом угла и радужки. Нарушение развития системы оттока внутриглазной жидкости приводит к глаукоме в 70% случаев.

↑ ДИАГНОСТИКА
Диагностика основана на данных соматического и офтальмологического обследования.
↑ Анамнез
Родители предъявляют жалобы на врождённое, чаще двустороннее помутнение роговицы у ребёнка.
↑ Инструментальные исследования
При биомикроскопии выявляют центральное помутнение роговицы, врождённую переднюю полярную катаракту, иридокорнеальные тяжи, сращения с полюсом люксированного в переднюю камеру хрусталика.
Офтальмоскопия затруднена.
При гониоскопии определяют частично или полностью закрытый передними периферическими синехиями угол, в УПК — мезенхимальную ткань.
В-сканирование и ультразвуковая биомикроскопия позволяют выявить степень патологических изменений передней камеры глаза: передние центральные и периферические синехии, эктопию хрусталика.
↑ Дифференциальная диагностика
Проводят с врождённой декомпенсированной глаукомой, сопровождаемой помутнением роговицы, врождёнными увеитами и кератитами.
↑ Показания к консультации других специалистов
Необходима консультация невропатолога и педиатра.
↑ Пример формулировки диагноза
Врождённая декомпенсированная глаукома, центральное помутнение роговицы, сращённое с тяжами радужки, люксация хрусталика в переднюю камеру, врождённая катаракта, синдром Петерса.
↑ ЛЕЧЕНИЕ
Лечение синдрома заключается в проведении кератопластики с реконструкцией передней камеры, при втором типе синдрома — с ленсэктомией. При развитии вторичной глаукомы показаны фистулизирующие операции. Исход кератопластики в большинстве случаев определяется степенью компенсации ВГД.
↑ Дальнейшее ведение
При необходимости показана пожизненная медикаментозная гипотензивная терапия или хирургическое лечение глаукомы и сопутствующей глазной патологии, а также терапия, направленная на улучшение трофики, 2 раза в год.
↑ ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Необходимость диспансерного наблюдения не реже 1 раза в 3 мес с регулярным контролем зрительных функций и ВГД.
↑ ПРОГНОЗ
Прогноз зависит от тяжести врождённой патологии. При синдроме Петерса типа 1 острота зрения зависит от степени помутнения роговицы, может быть снижена до сотых. При синдроме Петерса типа II наблюдают врождённую слепоту или слабовидение. Дальнейший прогноз зависит от стабилизации глаукомного процесса. Возможна инвалидность по зрению.
↑ Синдром Франк-Каменецкого синонимы
Врождённая семейная гипоплазия радужки.
↑ ОПРЕДЕЛЕНИЕ
Синдром Франк-Каменецкого — врождённая двусторонняя гипоплазия радужки, наследуемая по рецессивному, Х-сцепленному с полом типу. Ассоциирована с гониодисгенезом и формированием глаукомы.
↑ КОД ПО МКБ-10
Н40.5 Глаукома вторичная вследствие других болезней глаз.
Н21.2 Дегенерация радужной оболочки и цилиарного тела.
↑ ЭПИДЕМИОЛОГИЯ
Глаукома развивается, как правило, в 20-30 лет, при сочетании синдрома с мегалокорнеа— в возрасте до 5 лет. Глаукомой Франк-Каменецкого болеют только мужчины, женщины-носители патологического гена имеют микропризнаки заболевания: тенденцию к двухцветному окрашиванию радужки, истончение стромы частичный задний эмбриотоксон.
↑ ЭТИОЛОГИЯ
Заболевание наследственное, передаётся по Х-сцепленному рецессивному типу женщинами больным сыновьям, заболевание передаётся через поколение, сыновья больных отцов всегда здоровы, дочери больных отцов - носительницы патологического гена.
↑ ПАТОГЕНЕЗ
Во всех случаях выявляют признаки гониодисгенеза, что и вызывает нарушения оттока внутриглазной жидкости и развитие глаукомы. Глаукома протекает по типу открытоугольной. никогда не бывает острой декомпенсации ВГД.
↑ КЛИНИЧЕСКАЯ КАРТИНА ДИАГНОСТИКА
Синдром диагностируют по типичным изменениям радужки и данным анамнеза.
↑ Анамнез
В анамнезе выявляют признаки, подтверждающие наследование болезни по X-сцепленному с полом виду - заболевание передаётся через поколение, больше подвержены мальчики, сыновья больных отцов всегда здоровы, дочери больных отцов - носительницы патологического гена.
Жалобы появляются только в стадии глаукомы.
↑ Инструментальные исследования
При биомикроскопии в 40% случаев выявляют мегалокорнеа. С рождения определяют двустороннюю гипоплазию стромы с типичным двуцветным окрашиванием радужек (рис. 33-53): зрачковая зона резко утолщена, светло-серая, периферия в виде широкого контрастного кольца коричневого или сине-лилового цвета.
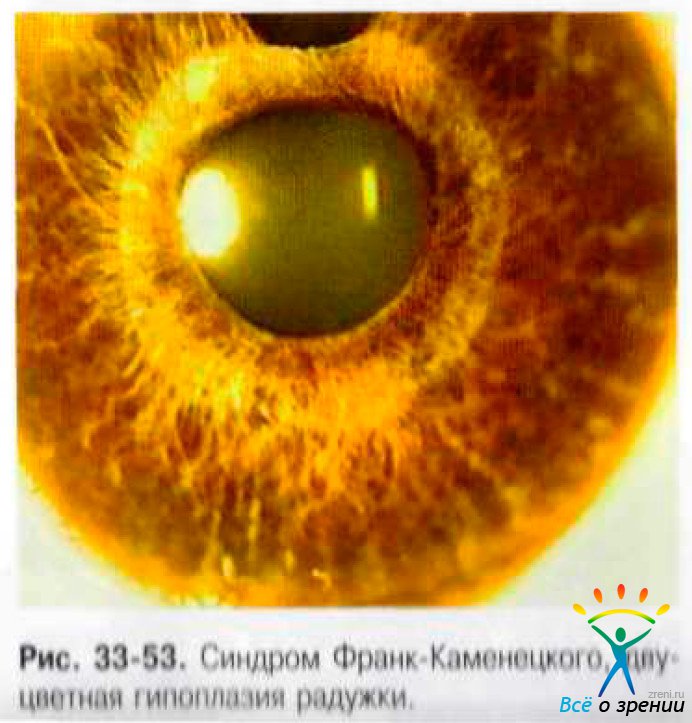
С возрастом возможно прогрессирование изменений радужки с формированием сквозных дырчатых треугольных разрывов основанием к лимбу, поликории и грубой деформации зрачка (рис. 33-54).

При офтальмоскопии выявляют нормальное глазное дно.
При гониоскопии находят: гониодисгенез в виде «зубчатого» и переднего крепления корня радужки, серую войлокоподобную ткань в проекции трабекулы, задний эмбриотоксон.
Ультразвуковая биомикроскопия и ОКТ радужки позволяют выявить степень её изменений: с рождения строма истончена вплоть до полного отсутствия в цилиарной зоне, пигментный листок превышает нормальные показатели в 2-3 раза.
↑ Дифференциальная диагностика
Синдром Франк Каменецкого необходимо дифференцировать с синдромом Ригера и с прогрессирующей эссенциальной мезодермальной атрофией.
↑ Показания к консультации других специалистов
Генетическое консультирование при планировании рождения детей.
↑ Пример формулировки диагноза
Юношеская декомпенсированная глаукома, синдром Франк Каменецкого.
↑ ЛЕЧЕНИЕ
Лазерное лечение неэффективно. Хирургическое лечение рассматривают как метод выбора, проводят фистулизирующие операции. Местные гипотензивные препараты назначают при необходимости после хирургического лечения.
↑ ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Необходимость диспансерного наблюдения не реже 1 раза в 3 мес с регулярным контролем зрительных функций и ВГД.
↑ Дальнейшее ведение
При необходимости показана пожизненная медикаментозная гипотензивная терапия или хирургическое лечение глаукомы и сопутствующей глазной патологии, а также терапия, направленная на улучшение трофики, 2 раза в год.
↑ ПРОГНОЗ
Зависит от наличия сопутствующей патологии. При сочетании с мегалокорнеа наблюдают раннее развитие и рефрактерное течение глаукомы, прогрессирующую дистрофию радужки, сопровождаемую снижением зрительных функций до слепоты и слабовидения к 30-40 годам жизни. При развитии глаукомы к возрасте 20 30 лет прогноз более благоприятный и зависит от степени компенсации ВГД.
↑ ВТОРИЧНАЯ ЗАКРЫТОУГОЛЬНАЯ ГЛАУКОМА С ПРОГРЕССИРУЮЩИМ ФОРМИРОВАНИЕМ ЭНДОТЕЛИАЛЬНОЙ МЕМБРАНЫ
↑ Иридокорнеальный эндотелиальный синдром
Иридокорнеальный эндотелиальный синдром — группа заболеваний с прогрессирующими изменениями корнеального эндотелия, избыточная пролиферация которого инициирует формирование периферических передних синехий и развитие вторичной закрытоугольной глаукомы.
↑ КОД ПО МКБ-10
Н40.5 Глаукома вторичная вследствие других болезней глаз.
Н21.2 Дегенерация радужной оболочки и цилиарного тела.
Н18.2 Другие отёки роговицы.
Н18.9 Болезнь роговицы неуточнённая.
↑ ЭПИДЕМИОЛОГИЯ
Начальные проявления в виде косметических дефектов радужки появляются в возрасте 20-50 лет, по данным некоторых авторов, от 0 до 60 лет. Процесс односторонний спорадический, не выявлено связи с другими глазными или системными заболеваниями. Чаще болеют женщины европеоидной расы.
↑ КЛАССИФИКАЦИЯ
Специфический характер мезодермальной атрофии радужки позволяет выделить три клинические формы иридокорнеального эндотелиального синдрома:
- синдром Чандлера;
- синдром Когана-Риза:
- прогрессирующая эссенциальная мезодермальная атрофия радужки. ЭТИОЛОГИЯ
Существует теория хронического воспаления герпесвирусной этиологии, и результате которого развиваются прогрессирующие патологические изменении эндотелия роговицы и его пролиферация на структуры передней камеры глаза.
↑ ПАТОГЕНЕЗ
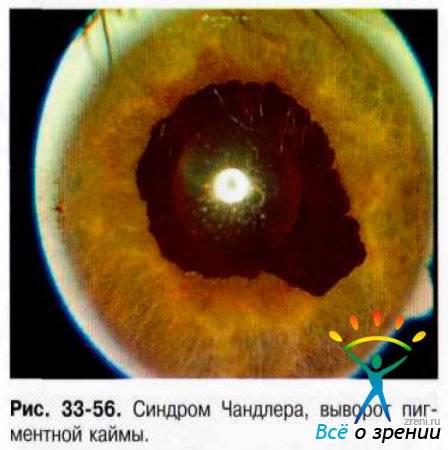
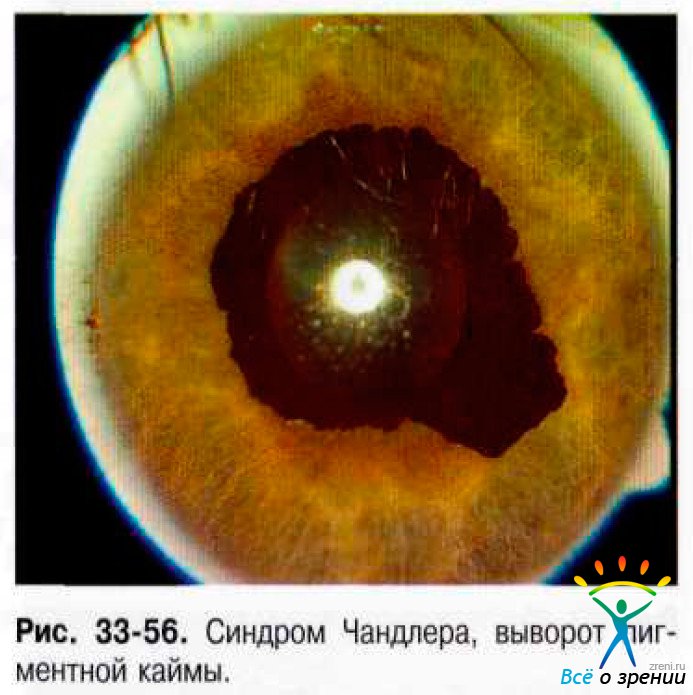
Изменённый корнеальный эндотелий с базальной мембраной распространяется с роговицы на трабекулярную часть УПК и переднюю поверхность радужки. Сокращение этой мембраны приводит к развитию периферической передней синехии в зонах ранее открытого угла и формированию глаукомы по типу закрытоугольной. Натяжение радужки сопровождают появление выворота пигментной каймы, эктопия зрачка, сквозные разрывы, узелки радужки.
↑ КЛИНИЧЕСКАЯ КАРТИНА
Пациенты предъявляют жалобы на боли в глазу, снижение, затуманивание зрения и косметические дефекты радужки, могут описывать «тёмные пятна» в глазу, которые соответствуют сквозным дефектам радужки.
↑ ДИАГНОСТИКА
Диагностика основана на данных офтальмологического обследования.
↑ Инструментальные исследования
При биомикроскопии выявляют диффузный отёк роговицы, изменения эндотелия по типу "чеканного серебра", своеобразные изменения радужки, соответствующие следующим вариантам течения иридокорнеального эндотелиального синдрома.
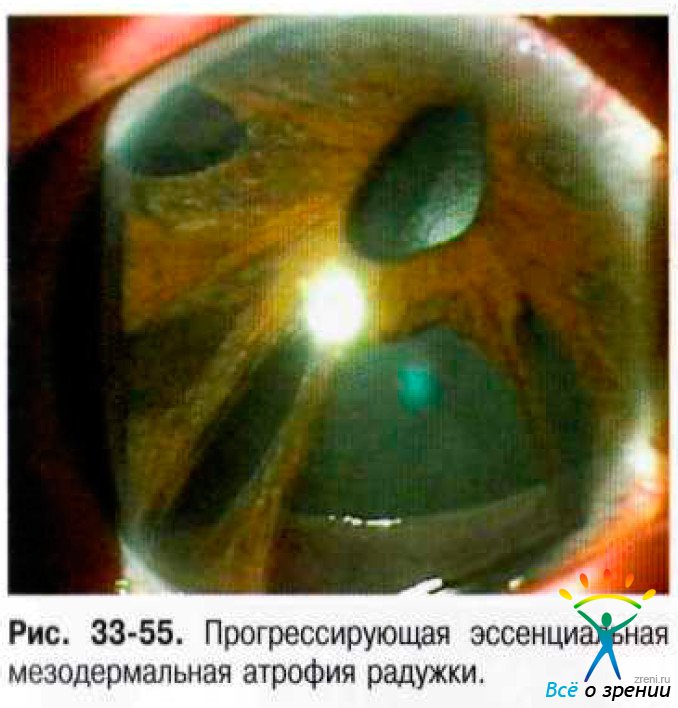
- Прогрессирующая эссенциальная мезодермальная атрофия радужки. Характерны выраженная атрофия радужки с прогрессирующим истончением стромы вплоть до образования дырчатых дефектов, эктопия зрачка, остатков стромы в виде тонких тяжей, отходящих от зрачкового пояса радужки (рис. 33-55).
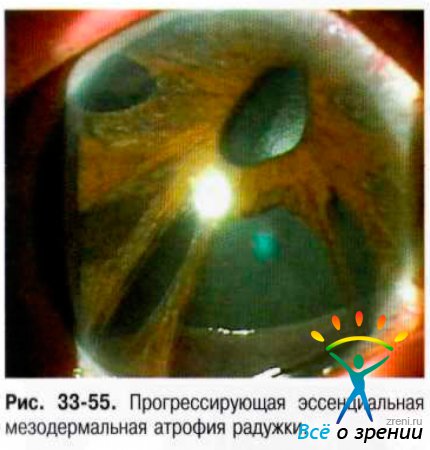
- Синдром Чандлера. Доминирующая черта этого варианта - отёк роговицы, часто при нормальном или умеренно повышенном ВГД. Выворот пигментной каймы (рис. 33-56).
- Синдром Когана Риза. Выворот пигментной каймы радужки при этом варианте течения заболевания сочетай с множественными узелковыми изменениями стромы радужки по типу невусов.
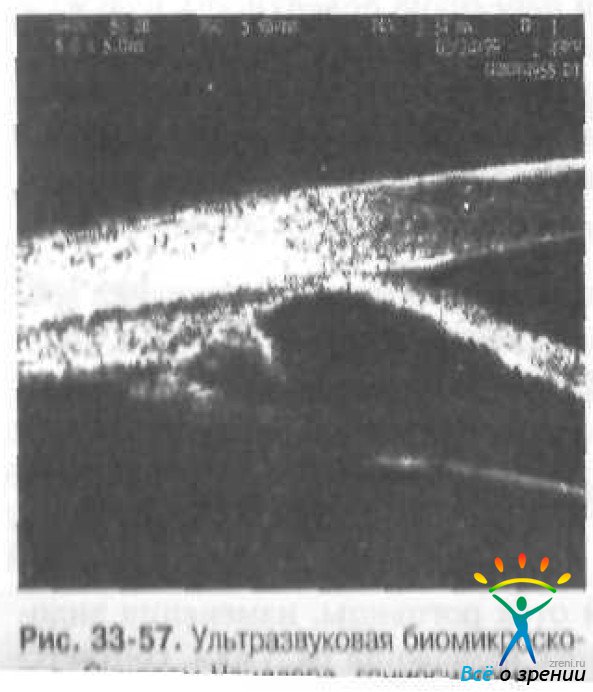
При гониоскопии выявляют характерный признак всех форм иридокорнеального эндотелиального синдрома — прогрессирующую высокую плоскостную переднюю периферическую синехию.
Ультразвуковая биомикроскопия позволяет диагностировать наличие и протяжённость гониосинехии (рис. 33-57).
ОКТ радужки выявляет наличие патологических изменений стромы: на её передней поверхности определяют плотную практически непрозрачную структуру, напоминающую рубцовую соединительную ткань.
Эндотелиальная зеркальная микроскопия выявляет полиморфизм, «тёмные поля» и уменьшение количества клеток эндотелия.
↑ Дифференциальная диагностика
Проводят с синдромом Франк—Каменецкого, синдромом Ригера. вторичными увеальными и посттравматическими глаукомами, изменениями радужки при её новообразованиях.
↑ Пример формулировки диагноза
Вторичная закрытоугольная декомпенсированная глаукома, кератопатия, прогрессирующая эссенциальная мезодермальная атрофия радужки.
↑ ЛЕЧЕНИЕ
Показаны фистулизирующие антиглаукомные операции с антиметаболитами, лазерная гониопунктура в послеоперационном периоде, когда снижение эффективности хирургического лечения связано с пролиферацией эндотелиальной мембраны на зону внутренней фистулы.
↑ ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Необходимо диспансерное наблюдение не реже 1 раза в 3 мес с регулярным контролем зрительных функций и ВГД.
↑ Дальнейшее ведение
Первичное хирургическое лечение, при необходимости — пожизненная медикаментозная гипотензивная терапия глаукомы, курсовая трофическая терапия 2 раза в год, при необходимости — кератопластическое лечение.
↑ ПРОГНОЗ
Недостаточно благоприятный при прогрессирующей эссенциальной мезодермальной атрофии радужки, которая сопровождается нарушением её диафрагмальной функции и снижением остроты зрения. В целом состояние зрительных функций определено степенью компенсации ВГД.
↑ ВТОРИЧНАЯ ОТКРЫТОУГОЛЬНАЯ ГЛАУКОМА, ОФТАЛЬМОЛОГИЧЕСКИ ОБУСЛОВЛЕННАЯ
↑ Синдром пигментной дисперсии
Синдром пигментной дисперсии — латентная стадия пигментной глаукомы, когда экзопигмента ещё недостаточно для формирования необратимых изменений трабекулы и формирования пигментной глаукомы.
↑ КОД ПО МКБ-10
Н40.1 Первичная открытоугольная глаукома.
↑ ЭПИДЕМИОЛОГИЯ
Синдром пигментной дисперсии начинается в 18-25 лет, пигментная глаукома — в 30-50 лет. Удельный вес составляет 1,5-2,0% от всех случаев глаукомы. Болеют преимущественно мужчины — миопы. С возрастом может происходить самопроизвольная ликвидация обратного зрачкового блока и вымывание пигмента, глаукому в таких случаях диагностируют как первичную хроническую открытоугольную глаукому.
↑ ЭТИОЛОГИЯ
Этиология синдрома не установлена. К факторам риска развития синдрома пигментной дисперсии относят: миопию, глубокую переднюю камеру, трабекулярное строение радужки, пролапс радужки у корня.
↑ ПАТОГЕНЕЗ
Согласно теории обратного зрачкового блока, при наличии вышеуказанных факторов риска в работающей как клапан радужке возникает патологический иридозонулярный контакт, в результате которого трение между радужкой и цинновыми связками приводит к механическому разрушению пигментного листка радужки. Гранулы меланина скапливаются в трабекуле, вызывая пре- и трабекулярную ретенцию, через 5-10 лет от начала синдрома пигментной дисперсии происходит переход синдрома пигментной дисперсии в пигментную глаукому.
↑ КЛИНИЧЕСКАЯ КАРТИНА
Жалобы выявляют только в стадии глаукомы во время острых эпизодов подъёма ВГД, спровоцированных расширением зрачка, физической нагрузкой, занятиями спортом, связанными с прыжками.
↑ ДИАГНОСТИКА
Диагностика основана на данных офтальмологического обследования.
↑ Инструментальные исследования
При биомикроскопии выявляют глубокую переднюю камеру (глубиной 3,5-5,0 мм), зоны трансиллюминации радужки в проекции участков разрушенного пигментного эпителия (рис. 33-58).

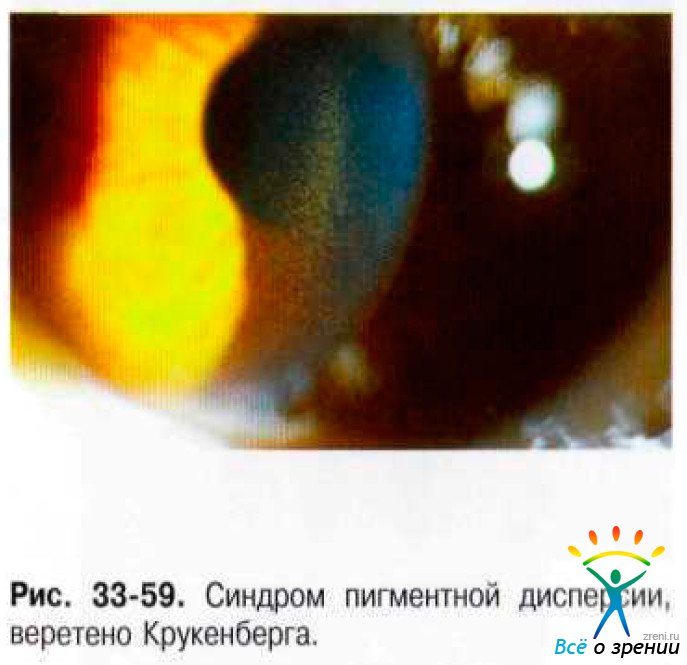
Дисперсия пигмента на всех структурах переднего отрезка глаза: на эндотелии роговицы в виде вертикального веретена Крукенберга (рис. 33-59), по корнеальному эндотелию у лимба, на передней поверхности радужки, по экватору хрусталика, на передней гиалоидной мембране.
При офтальмоскопии в далеко зашедшей стадии пигментной дисперсии выявляют гранулы меланина в СТ по ходу клокетова канала и перипапиллярно, в 12% случаев диагностируют периферические дегенерации сетчатки.
При гониоскопии находят всегда открытым УПК, канавообразный профиль, степень пигментации 3-4.
В стадии манифестации синдрома пигментной дисперсии нестабильное состояние гидродинамики глаза подтверждают положительной нагрузочной пробой с мидриатиками (повышение ВГД после инстилляции тропикамида более 5 мм рт. ст.).
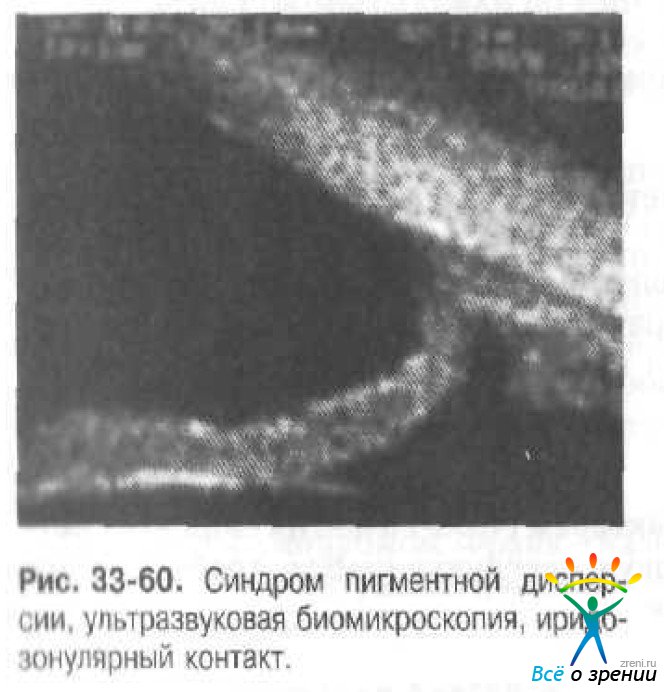
Ультразвуковая биомикроскопическая диагностика иридозонулярного контакта (рис. 33-60).
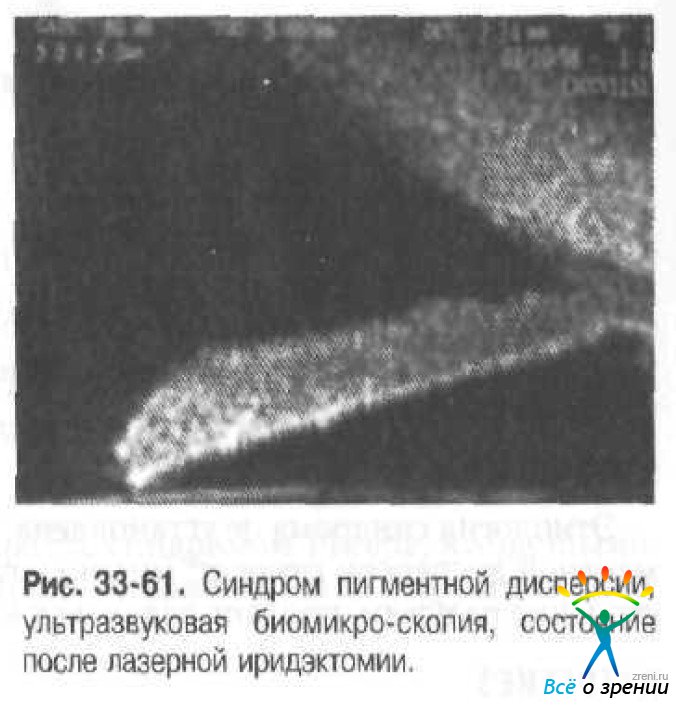
ОКТ: диагностика дефектов пигментного эпителия радужки, динамика его регенерации после лазерной иридэктомии (рис. 33-61).
↑ Дифференциальная диагностика
Проводят с первичной хронической открытоугольной глаукомой и псевдоэксфолиативной открытоугольной глаукомой.
↑ ЛЕЧЕНИЕ
Препараты, расширяющие зрачок, могут приводить к дополнительному выбросу гранул меланина и повышению ВГД.
Медикаментозно ликвидировать обратный зрачковый блок возможно назначением миотиков. Методом выбора при лечении синдрома пигментной дисперсии считают периферическую лазерную иридэктомию. Лазерная гониотрабекулопластика при четвёртой степени пигментации трабекулы малоэффективна. При развитии пигментной глаукомы показаны фистулизирующие операции.
↑ ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Необходимость диспансерного наблюдения не реже 1 раза в 3 мес с регулярным контролем зрительных функций и ВГД.
↑ Дальнейшее ведение
Первичное лазерное лечение, при необходимости показана пожизненная медикаментозная гипотензивная терапия или хирургическое лечение глаукомы. Курсовая трофическая терапия 2 раза в год.
---
Статья из книги: Офтальмология. Национальное руководство | Аветисов С.Э.

Комментариев 0