Сетчатка ? Часть 2


Содержание:
Описание
↑ Болезни сетчатки
Болезни сетчатки очень разнообразны. Они обусловлены воздействием различных факторов, приводящих к патологоанатомическим и патологофизиологическим изменениям, что в свою очередь определяет нарушения зрительных функций и наличие характерных симптомов. Среди заболеваний сетчатки выделяют наследственные и врожденные дистрофии, болезни, обусловленные инфекциями, паразитами и аллергическими агентами, сосудистыми нарушениями и опухолями. Несмотря на разнообразие заболеваний сетчатки, патологоанатомические и патофизиологические проявления могут быть сходными при разных нозологических формах.
К патологическим процессам, наблюдаемым в сетчатке, относят дистрофии, которые могут быть генетически детерминированными или вторичными, воспаление и отек, ишемию и некроз, кровоизлияния, отложение твердых или мягких экссудатов и липидов, ретиношизис и отслойку сетчатки, фиброз, пролиферацию и образование неоваскулярных мембран, гиперплазию и гипоплазию пигментного эпителия, опухоли, ангиоидные полосы. Все эти процессы можно выявить при офтальмоскопии глазного дна.
↑ Наследственные генерализованные дистрофии
Фоторецепторные дистрофии сетчатки различаются типом наследования, характером нарушения зрительных функций и картиной глазного дна в зависимости от первичной локализации патологического процесса в различных структурах: мембране Бруха, пигментном эпителии сетчатки, в комплексе пигментный эпителий — фоторецепторы, фоторецепторах и внутренних слоях сетчатки. Дистрофии сетчатки как центральной, так и периферической локализации могут быть следствием мутации гена родопсина и перифирина. При этом симптомом, объединяющим эти заболевания, является стационарная ночная слепота.
К настоящему времени известно 11 хромосомных районов, которые содержат гены, мутации которых являются причиной развития пигментного ретинита, и для каждого генетического типа пигментного ретинита характерны аллельная и неаллельная разновидности.
Пигментный ретинит (пигментное перерождение сетчатки, тапеторетинальная дегенерация) — заболевание, характеризующееся поражением пигментного эпителия и фоторецепторов с разными типами наследования: аутосомно-доминантным, аутосомно-рецессивным или сцепленным с полом. Возникает в результате образования дефектов генетического кода, следствием чего является аномальный состав специфических белков. Течение заболевания при разных типах наследования имеет некоторые особенности. Ген родопсина — первый идентифицированный ген, мутации которого являются причиной развития пигментного ретинита с аутосомно-доминантным типом наследования.
Заболевание проявляется в раннем детском возрасте и характеризуется триадой симптомов: типичными пигментными очагами (рис. 15.7,а)
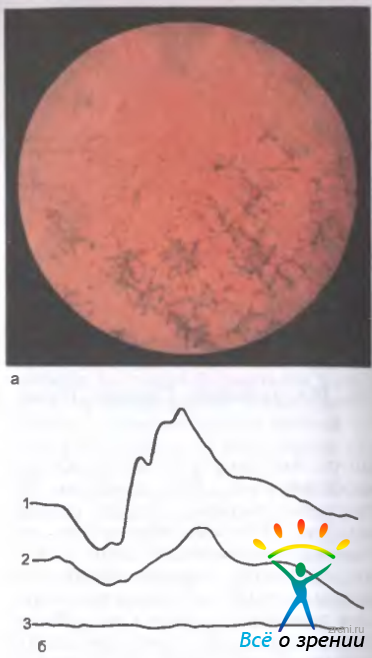
Рис. 15.7. Пигментный ретинит. а — глазное дно больной пигментным ретинитом; б — ЭРГ у членов семьи больной: 1 — нормальная у здорового мужа, 2 — субнормальная у сына — носителя патологического гена, 3 — нерегистрируемая у больной.
на средней периферии глазного дна и по ходу венул (их называют костными тельцами), восковидной бледностью диска зрительного нерва, сужением артериол.
У больных с пигментным ретинитом со временем могут развиться пигментные изменения в макулярной области в связи с дегенерацией фоторецепторов, что сопровождается снижением остроты зрения, задней отслойкой стекловидного тела и отложением в нем нежного пигмента. Возможно возникновение макулярного отека, обусловленного проникновением жидкости из хориоидеи через пигментный эпителий, а по мере развития процесса — преретинального макулярного фиброза. У больных с пигментным ретинитом с большей частотой, чем в общей популяции, встречаются друзы диска зрительного нерва, задняя субкапсулярная катаракта, открытоугольная глаукома, кератоконус и миопия. Хориоидея долго остается интактной и вовлекается в процесс только в поздних стадиях заболевания.
В связи с поражением палочковой системы возникает ночная слепота, или никталопия. Темновая адаптация нарушена уже в начальной стадии заболевания, порог световой чувствительности повышен как в палочковой, так и в колбочковой части.
Функциональные методы исследования позволяют выявить прогрессирующие изменения в фоторецепторах. При периметрии на средней периферии (30—50°) обнаруживают кольцевые полные и неполные скотомы, которые расширяются к периферии и центру. В поздней стадии заболевания поле зрения концентрически суживается до 10°, сохраняется лишь центральное трубчатое зрение.
Отсутствие или резкое снижение общей ЭРГ является патогномоничным признаком пигментного ретинита (рис. 15.7,б).
Локальная ЭРГ долго остается нормальной, а изменения наступают при вовлечении в патологический процесс колбочковой системы макулярной области. У носителей патологического гена отмечаются редуцированная ЭРГ и удлиненный латентный период b-волны ЭРГ, несмотря на нормальное глазное дно.
Атипичные формы пигментного ретинита. К другим формам пигментного ретинита относятся пигментный инвертированный ретинит (центральная форма), пигментный ретинит без пигмента, белоточечный пигментный ретинит и псевдопигментный ретинит. Каждая из этих форм имеет характерную офтальмоскопическую картину и электроретинографическую симптоматику.
Пигментный инвертированный ретинит (центральная форма). В отличие от типичной формы пигментного ретинита заболевание начинается в макулярной области и поражения колбочковой системы более значительны, чем палочковой. В первую очередь снижается центральное и цветовое зрение, появляется фотофобия (светобоязнь). В макулярной области отмечаются характерные пигментные изменения, которые могут сочетаться с дистрофическими изменениями на периферии. В таких случаях одним из основных симптомов является отсутствие дневного зрения. В поле зрения центральная скотома, на ЭРГ значительно редуцированы колбочковые компоненты по сравнению с палочковыми.
Пигментный ретинит без пигмента. Название связано с отсутствием характерных для пигментного ретинита пигментных отложений в виде костных телец при наличии симптомов, сходных с проявлениями пигментного ретинита, и нерегистрируемой ЭРГ.
Белоточечный пигментный ретинит. Характерным офтальмоскопическим признаком являются множественные белые точечные пятна по всему глазному дну с сопутствующими пигментными изменениями ("ткань, изъеденная молью") или без них. Функциональные симптомы сходны с проявлениями пигментного ретинита. Заболевание необходимо дифференцировать от стационарной врожденной ночной слепоты и белоточечного глазного дна (fundus albipunctatus).
Псевдопигментный ретинит — ненаследственное заболевание. Причиной его возникновения могут быть воспалительные процессы в сетчатке и хориоидее, побочное действие лекарственных препаратов (тиоридазин, меллирил, хлороквин, дефероксамин, клофазамин и др.), состояние после травмы, отслойки сетчатки и т. д. На глазном дне выявляют изменения, сходные с таковыми при пигментном ретините. Основным отличительным симптомом является нормальная или незначительно сниженная ЭРГ. При этой форме никогда не бывает нерегистрируемой или резко сниженной ЭРГ.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
В настоящее время патогенетически обоснованного лечения пигментного ретинита не существует. Заместительная или стимулирующая терапия неэффективна. Больным с пигментным ретинитом рекомендуют носить темные защитные очки для предотвращения повреждающего действия света, подбор максимальной очковой коррекции остроты зрения, назначают симптоматическое лечение: при макулярном отеке — системное и местное использование диуретиков (ингибиторов карбоангидразы), например диакарба, диамокса (ацетазоламид); при наличии помутнений хрусталика хирургическое лечение катаракты для улучшения остроты зрения, при наличии неоваскуляризации для профилактики осложнений проводят фотокоагуляцию сосудов, назначают сосудистые препараты. Больные, их родственники и дети должны проходить генетическое консультирование, исследование других органов и систем для исключения синдромных поражений и других болезней.
Идентификация патологического гена и его мутаций является основой понимания патогенеза заболевания, прогнозирования течения процесса и поиска путей рациональной терапии. В настоящее время в эксперименте предпринимаются попытки трансплантации клеток пигментного эпителия и нейрональных клеток сетчатки от недельного зародыша. Новый многообещающий подход к лечению пигментного ретинита связан с генной терапией, основанной на субретинальном введении аденовируса, с содержанием внутри капсулы здоровых мини-хромосом. Ученые предполагают, что вирусы, проникая в клетки пигментного эпителия, способствуют замещению мутированных генов.
Генерализованная наследственная дистрофия сетчатки, связанная с системными заболеваниями и нарушениями метаболизма. Существует множество системных расстройств, которые сочетаются с атипичными формами пигментного ретинита. К настоящему времени известно около 100 заболеваний с различными глазными нарушениями, обусловленных нарушением метаболизма липидов, углеводов, протеинов. Недостаточность внутриклеточных энзимов приводит к мутациям генов, что определяет различную генетическую патологию, в том числе исчезновение или дистрофию фоторецепторных клеток.
К специфическим системным заболеваниям, сочетающимся с пигментным ретинитом, относят нарушения метаболизма углеводов (мукополисахароидозы), липидов (муколипидозы, фукозидоз, сероидные липофусцинозы), липопротеинов и протеинов, поражения центральной нервной системы, синдромы Ушера, Лоренса—Муна—Барде—Бидля и др.
Врожденный амавроз Лебера — наиболее тяжелое проявление пигментного ретинита (генерализованная форма), наблюдающаяся с рождения. Основные симптомы: отсутствие центрального зрения, нерегистрируемая или резко субнормальная ЭРГ, нистагм. Диагностика врожденного амавроза Лебера очень сложна, поскольку выявляемые у больных симптомы: косоглазие, кератоконус, гиперметропия высокой степени, неврологические и нейромышечные нарушения, снижение слуха, умственная отсталость, могут быть и при других системных заболеваниях.
Чаще отмечается аутосомно-рецессивный тип передачи заболевания, и его еще недавно связывали с двумя различными генами, в том числе с мутацией гена родопсина. В настоящее время амавроз Лебера рассматривают как гетерогенную группу нарушений, при которых поражаются палочки и колбочки.
При этом заболевании дети либо рождаются слепыми, либо теряют зрение в возрасте около 10 лет. В течение первых 3—4 мес жизни большинство родителей отмечают у них отсутствие фиксации предметов и реакции на свет, типичные глазные симптомы, характерные для детей, рожденных слепыми: блуждающий взор и нистагм, которые отмечаются уже в первые месяцы жизни. У младенцев глазное дно может выглядеть нормальным, однако со временем патологические изменения обязательно появляются. Глазные симптомы включают различные пигментные изменения в заднем полюсе глаза от гиперпигментированных до непигментированных очагов по типу соль с перцем, миграцию пигмента в сетчатке и пигментные скопления, атрофию пигментного эпителия и капилляров хориоидеи, реже — множественные неравномерно расположенные желтовато-белые пятна на периферии и средней периферии сетчатки. Пигментные отложения в виде костных телец на периферии глазного дна обнаруживают почти у всех больных с амаврозом Лебера в возрасте 8—10 лет, однако эти отложения могут быть очень мелкими, напоминают сыпь при краснухе. Диск зрительного нерва, как правило, бледный, сосуды сетчатки сужены. Изменения на глазном дне обычно прогрессируют, но в отличие от типичного пигментного ретинита функциональные изменения (острота зрения, поле зрения, ЭРГ) обычно остаются такими же, как при первоначальном осмотре. Со временем, после 15 лет, у больных с амаврозом Лебера может развиться кератоконус.
При микроскопическом исследовании выявляют субретинальные включения, состоящие из отторгнутых наружных сегментов фоторецепторов и макрофагов, на участках, соответствующих офтальмоскопически видимым белым пятнам. Наружные сегменты палочек отсутствуют, сохраняется небольшое количество измененных колбочек, другие клеточные элементы представляют собой недифференцированные фоторецепторы и эмбриональные клетки пигментного эпителия.
Причиной заболевания является отсутствие дифференциации клеток пигментного эпителия и фоторецепторов.
Дифференциальную диагностику проводят с доминантной атрофией зрительного нерва, при которой прогноз относительно зрения значительно лучше, а также с подобными изменениями при краснухе и врожденном сифилисе.
Основным симптомом врожденного амавроза Лебера, на котором основана дифференциальная диагностика, является либо нерегистрируемая, либо резко субнормальная ЭРГ, в то время как при атрофии зрительного нерва, сифилисе и краснухе ЭРГ либо нормальная, либо субнормальная. При врожденном амаврозе Лебера возможны различные распространенные неврологические и нейродегенеративные нарушения, церебральная атрофия, отмечаются задержка умственного развития, болезни почек. Однако интеллектуальное развитие детей с врожденным амаврозом Лебера может быть нормальным.
Фоторецепторные дисфункции. Дисфункции палочковой системы. Врожденная стационарная ночная слепота. Врожденная стационарная ночная слепота, или никталопия (отсутствие ночного зрения), — непрогрессирующее заболевание, причиной которого является дисфункция палочковой системы. При гистологическом исследовании структурных изменений в фоторецепторах не выявляют. Результаты электрофизиологических исследований подтверждают наличие первичного дефекта в наружном плексиформном (синаптическом) слое, так как нормальный палочковый сигнал не достигает биполярных клеток. Выделяют различные типы стационарной ночной слепоты, которые дифференцируются по ЭРГ.
Врожденная стационарная ночная слепота с нормальным глазным дном характеризуется разными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и сцепленным с Х-хромосомой.
Врожденная стационарная ночная слепота с изменением глазного дна. К этой форме заболевания относится болезнь Огуши — заболевание с аутосомно-рецессивным типом наследования, которое отличается от стационарной врожденной ночной слепоты изменениями на глазном дне, проявляющимися желтоватым металлическим блеском, более выраженным в заднем полюсе. Макулярная область и сосуды на этом фоне выглядят рельефно. После 3 ч темновой адаптации глазное дно становится нормальным (феномен Мицуо). После световой адаптации глазное дно вновь медленно приобретает металлический блеск. При исследовании темновой адаптации выявляют заметное удлинение палочкового порога при нормальной колбочковой адаптации. Концентрация и кинетика родопсина в норме.
Белоточечное глазное дно (fundus albipunctatus) сравнивают со звездным небом ночью, поскольку на средней периферии глазного дна и в макулярной области регулярно расположены мириады беловатых мелких нежных пятнышек (рис. 15.8).
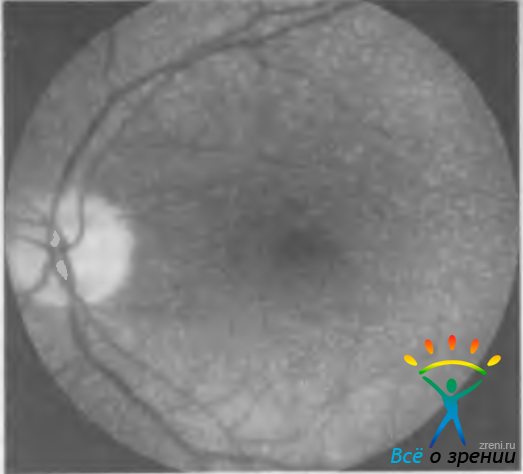
Рис. 15.8. Белоточечное глазное дно.
Заболевание с аутосомно-рецессивным типом наследования. На ФАГ выявляют фокальные области гиперфлюоресценции не связанные с белыми пятнами, которые на ангиограммах не видны.
В отличие от других форм стационарной ночной слепоты при белоточечном глазном дне отмечено замедление регенерации зрительного пигмента как в палочках, так и в колбочках. Амплитуда фотопических и скотопических а- и b-волн ЭРГ снижена при стандартных условиях регистрации. После нескольких часов темновой адаптации скотопический ответ ЭРГ медленно возвращается к норме.
Дисфункции колбочковой системы (синдром колбочковой дисфункции) проявляются нарушением цветовосприятия или полной ахромазией.
↑ Наследственные периферические дистрофии сетчатки
При этих формах дистрофии поражается оптически недеятельная часть сетчатки около зубчатой линии. В патологический процесс часто вовлекаются не только сетчатка и Хориоидея, но и стекловидное тело, в связи с чем они получили название "периферические витреохориоретинальные дистрофии".
Х-хромосомный ювенильный ретиношизис относится к наследственным витреоретинальным дегенерациям, сцепленным с полом. Зрение снижается в первой декаде жизни. Болеют мужчины. Ген RS1, ответственный за развитие Х-хромосомного ретиношизиса, локализуется на коротком плече 22-й хромосомы.
Расслоение сетчатки — основной клинический признак заболевания (см. рис. 15.15).
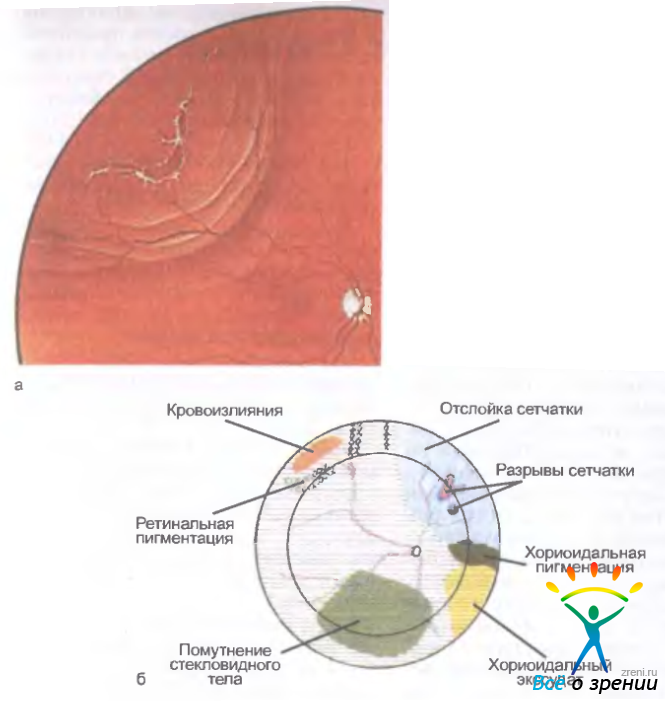
Рис. 15.15. Отслойка сетчатки. а — регматогенная отслойка сетчатки с разрывами; б — ретинальная карта с использованием международного цветового кода
Оно возникает в слое нервных волокон сетчатки. Предполагают, что ретиношизис является результатом нарушения функции опорных мюллеровских клеток. Ретиношизис сопровождается дистрофическими изменениями сетчатки, представленными участками золотисто-серебристого цвета; белые древовидные структуры образуются аномальными сосудами, проницаемость стенок которых повышена. На периферии часто формируются гигантские кисты сетчатки, окруженные пигментом. Эта форма заболевания, называемая буллезной, обычно наблюдается у детей раннего возраста и сочетается с косоглазием и нистагмом. Кисты сетчатки могут самопроизвольно спадаться. При прогрессировании ретиношизиса развиваются глиальная пролиферация, неоваскуляризация сетчатки, возможны множественные аркоподобные разрывы, гемофтальм или кровоизлияния в полость кист. В стекловидном теле определяются фиброзные тяжи, аваскулярные или васкулярные мембраны и вакуоли. Вследствие сращения тяжей с сетчаткой возникает натяжение (тракция), что приводит к возникновению тракционных разрывов сетчатки и ее отслойке. В макулярной области наблюдаются звездоподобные складки или радиальные линии в виде звезды ("спицы в колесе"). Острота зрения значительно снижена.
Ведущую роль в диагностике играют офтальмоскопическая картина заболевания и ЭРГ, которая резко субнормальна.
Лечение: лазерная коагуляция сетчатки и хирургическое лечение. При отслойке сетчатки производят витрэктомию, интравитреальную тампонаду перфторуглеродами или силиконовым маслом, экстрасклеральное пломбирование.
Болезнь Гольдмана — Фавре — прогрессирующая витреоретинальная дистрофия с аутосомно-рецессивным типом наследования, которая характеризуется сочетанием пигментного ретинита с костными тельцами, ретиношизисом (центральным и периферическим) и изменениями в стекловидном теле (дегенерация с формированием мембран). Нередко наблюдается осложненная катаракта. Частым осложнением является отслойка сетчатки.
Функциональные симптомы соответствуют клиническим проявлениям заболевания. Плохое сумеречное зрение и ночная слепота отмечаются уже в возрасте 5—10 лет. Острота зрения снижена, наблюдаются кольцевые скотомы или концентрическое сужение поля зрения. Темновая адаптация нарушена. Одним из основных симптомов является нерегистрируемая или резко субнормальная ЭРГ. Эффективных методов лечения в настоящее время нет. Назначают препараты, улучшающие микроциркуляцию и обменные процессы в сетчатке. При отслойке сетчатки производят хирургическое лечение.
Болезнь Вагнера также относится к витреоретинальным дистрофиям с аутосомно-доминантным типом наследования. Ген, ответственный за развитие болезни Вагнера, локализуется на длинном плече 5-й хромосомы. Основные клинические симптомы заболевания — наличие миопии, часто высокой степени, и преретинальных мембран при "оптически пустом" стекловидном теле в сочетании с ретиношизисом, дистрофией сетчатки и пигментного эпителия. Уже в возрасте 10—20 лет выявляют помутнения хрусталика, которые быстро прогрессируют; часто наблюдаются вторичная глаукома и отслойка сетчатки.
Диагноз устанавливают на основании семейного анамнеза, результатов биомикро- и офтальмоскопии, периметрии, электроретинографии и флюоресцентной ангиографии. При периметрии выявляют концентрическое сужение поля зрения, реже — кольцевидную скотому. ЭРГ резко субнормальная.
↑ Наследственные центральные дистрофии сетчатки
Центральные (макулярные) дистрофии — это заболевания, локализованные в центральном отделе сетчатки, характеризующиеся прогрессирующим течением, типичной офтальмоскопической картиной и сходной функциональной симптоматикой: снижение центрального зрения, нарушение цветового зрения, снижение колбочковых компонентов ЭРГ.
К наследственным наиболее распространенным макулярным дистрофиям с изменениями в пигментном эпителии сетчатки и фоторецепторах относят
- болезнь Штаргардта,
- желтопятнистое глазное дно,
- вителлиформную дистрофию Беста.
Болезнь Штаргардта и желтопятнистое глазное дно (желтопятнистая дистрофия). Болезнь Штаргардта — дистрофия макулярной области сетчатки, которая начинается в пигментном эпителии и проявляется двусторонним снижением остроты зрения в возрасте 10—20 лет. Заболевание описано К. Штаргардтом еще в начале XX в. как наследственное заболевание макулярной области с полиморфной офтальмоскопической картиной: "битая бронза", "бычий глаз", атрофия хориоидеи и др.
Путем позиционного клонирования определен основной локус гена для болезни Штаргардта, экспрессирующегося в фоторецепторах, который был назван ABCR. При аутосомно-доминантном типе наследования болезни Штаргардта установлена локализация мутированных генов в хромосомах 13q и 6ql4.
Результаты генетических исследований, проведенных в последние годы, свидетельствуют о том, что, несмотря на различия в клинической картине, пигментный ретинит, болезнь Штаргардта, желтопятнистое глазное дно и макулярная дегенерация, связанная с возрастом, являются аллельными нарушениями локуса ABCR.
Феномен "бычий глаз" офтальмоскопически виден как темный центр, окруженный широким кольцом гипопигментации, за которым обычно следует кольцо гиперпигментации (рис. 15.9, а).
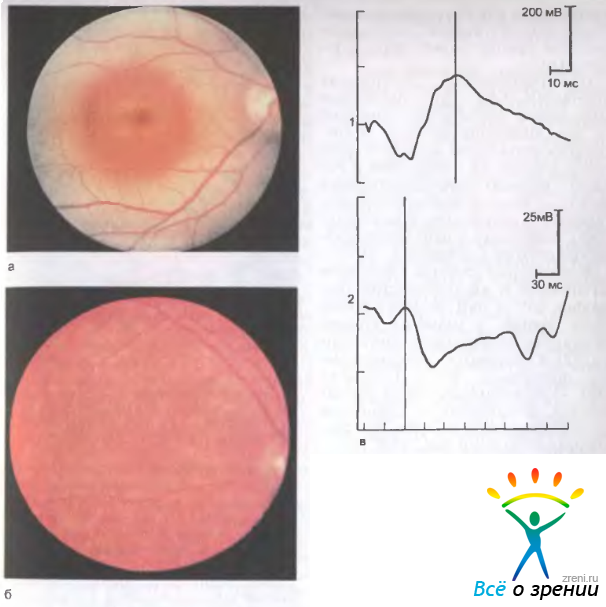
Рис. 15.9. Болезнь Штаргардта. а — глазное дно типа бычий глаз; б — желтопятнистое глазное дно; в — ЭРГ при болезни Штаргардта: 1 — общая нормальная, 2 — локальная субнормальная.
На ФАГ при типичном феномене "бычьего глаза" на нормальном фоне выявляются зоны отсутствия флюоресценции или гипофлюоресценции с видимыми хориокапиллярами. Гистологически отмечаются увеличение количества пигмента в центральной зоне глазного дна, атрофия прилежащего пигментного эпителия сетчатки и комбинация атрофии и гипертрофии пигментного эпителия. Отсутствие флюоресценции в макулярной области объясняется накоплением в пигментном эпителии сетчатки липофусцина, который является экраном для флюоресцеина. Липофусцин, кроме того, ослабляет окислительную функцию лизосом и увеличивает pH клеток пигментного эпителия сетчатки, что приводит к потере их мембранной целости.
Встречается редкая форма желтопятнистой дистрофии без изменений в макулярной области. В этом случае между макулой и экватором видны множественные желтоватые пятна различной формы: округлые, овальные, удлиненные, которые могут сливаться или располагаться отдельно друг от друга (рис. 15.9,б). Со временем цвет, форма и размеры этих пятен могут изменяться; изменяется и картина на ФАГ: участки с гиперфлюоресценцией становятся гипофлюоресцирующими, что соответствует атрофии пигментного эпителия сетчатки.
У всех больных с болезнью Штаргардта выявляют относительные или абсолютные центральные скотомы разной величины в зависимости от распространения процесса. При желтопятнистой дистрофии поле зрения может быть в норме при отсутствии изменений в макулярной области.
У большинства пациентов отмечается изменение цветового зрения по типу дейтеранопии, красно-зеленой дисхромазии или более выраженные. При желтопятнистой дистрофии цветовое зрение может быть нормальным.
Пространственная контрастная чувствительность при дистрофии Штаргардта значительно изменена во всем диапазоне частот со значительным снижением в области средних и полным ее отсутствием в области высоких пространственных частот — "паттерн колбочковой дистрофии". Контрастная чувствительность отсутствует в центральной области сетчатки в пределах 6—10°.
В начальных стадиях дистрофии Штаргардта и желтопятнистой дистрофии ЭРГ и ЭОГ остаются в пределах нормы, в развитых стадиях снижаются колбочковые компоненты ЭРГ и становятся субнормальными показатели ЭОГ. Локальная ЭРГ — субнормальна уже в ранних стадиях заболевания и становится нерегистрируемой по мере прогрессирования болезни (рис. 15.9, в).
Дифференциальную диагностику болезни Штаргардта следует проводить с доминантной прогрессирующей фовеальной дистрофией, колбочковой, колбочко-палочковой и палочко-колбочковой дистрофией, ювенильным ретиношизисом, вителлиформной макулярной дистрофией, приобретенными лекарственными дистрофиями (например, при хлороквиновой ретинопатии), при тяжелом токсикозе беременности.
Патогенетически обоснованного лечения не существует. Рекомендуется ношение солнцезащитных очков для предотвращения повреждающего действия света.
Желточная (вителлиформная) макулярная дистрофия Бестa. Болезнь Беста — редко наблюдаемая двусторонняя дистрофия сетчатки в макулярной области, имеющая вид круглого желтоватого очага, похожего на свежий яичный желток, диаметром от 0,3 до 3 диаметров диска зрительного нерва (рис. 15.10).
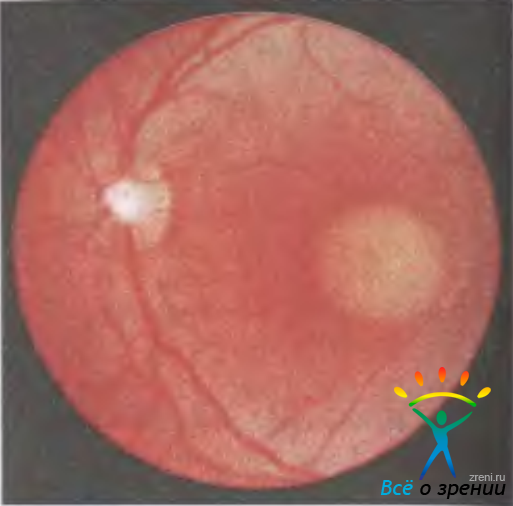
Рис. 15.10. Желточная вителлиформная дистрофия Беста.
Ген, ответственный за развитие болезни Беста, локализуется на длинном плече 11-й хромосомы (11q13). Тип наследования болезни Беста — аутосомно-доминантный.
В гистологических исследованиях установлено, что между клетками пигментного эпителия и нейроэпителия кумулируются гранулы вещества, подобного липофусцину, в субретинальном пространстве и в хориоидее — макрофаги, нарушается структура мембраны Бруха, во внутренних сегментах фоторецепторов накапливаются кислые мукополисахариды. Со временем развивается дегенерация наружных сегментов фоторецепторов.
Течение заболевания обычно бессимптомное, его выявляют случайно при осмотре детей в возрасте 5— 15 лет. Изредка пациенты предъявляют жалобы на затуманивание зрения, затруднения при чтении текстов с мелким шрифтом, метаморфопсии. Острота зрения варьирует в зависимости от стадии болезни от 0,02 до 1,0. Изменения в большинстве случаев асимметричные, двусторонние.
В зависимости от офтальмоскопических проявлений выделяют четыре стадии заболевания, хотя развитие макулярных изменений не всегда проходит через все стадии.
- Стадия I — минимальные нарушения пигментации в виде мелких желтых очажков в макуле;
- стадия II — классическая вигеллиформная киста в макуле;
- стадия III — разрыв кисты и различные фазы резорбции ее содержимого;
- стадия IV — формирование фиброглиального рубца с субретинальной неоваскуляризацией или без нее.
Диагноз устанавливают на основании результатов офтальмоскопии, флюоресцентной ангиографии, электроретинографии и электроокулографии. Помощь в диагностике может оказать обследование других членов семьи.
На ФАГ в I стадии заболевания отмечается локальная гиперфлюоресценция в зонах атрофии пигментного эпителия, во II стадии — отсутствие флюоресценции в области кисты. После разрыва кисты определяются гиперфлюоресценция в ее верхней половине и "блок" флюоресценции в нижней. После резорбции содержимого кисты в макуле выявляют окончатые дефекты.
Патогномоничный признак болезни Беста — патологическая ЭОГ. Общая и локальная ЭРГ не изменяются. В III—IV стадиях болезни выявляют центральную скотому в поле зрения.
Патогенетически обоснованного лечения не существует. В случае формирования субретинальной неоваскулярной мембраны может быть проведена лазерная фотокоагуляция.
Желточная вителлиформная макулодистрофия взрослых. В отличие от болезни Беста фовеолярные изменения развиваются в зрелом возрасте, имеют меньшие размеры и не прогрессируют. ЭОГ, как правило, не изменена.
Инволюционная макулярная дистрофия сетчатки (синонимы: возрастная, сенильная, центральная хориоретинальная дистрофия, макулярная дистрофия, связанная с возрастом; англ. Age-related macular dystrophy — АМД) является основной причиной снижения зрения у людей старше 50 лет. Это генетически обусловленное заболевание с первичной локализацией патологического процесса в пигментном эпителии сетчатки, мембране Бруха и хориокапиллярах макулярной области.
Офтальмоскопически выделяют следующие признаки: друзы (узелковые утолщения базальной мембраны пигментного эпителия сетчатки), пигментную (географическую) эпителиальную атрофию или гиперпигментацию, отслойку пигментного эпителия, субретинальные экссудаты (желтая экссудативная отслойка), кровоизлияния, фиброваскулярные рубцы, хориоидальную неоваскулярную мембрану, кровоизлияния в стекловидное тело (рис. 15.11).
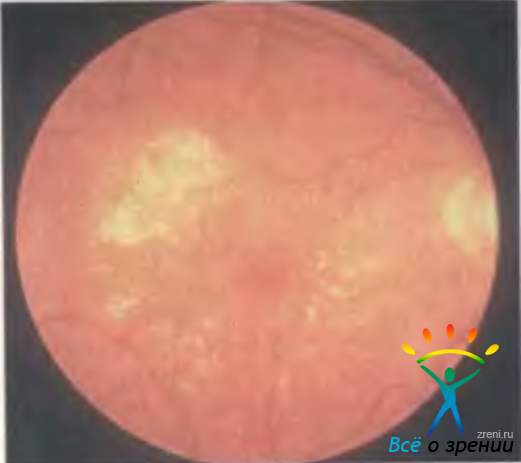
Рис. 15.11. Макулярная дегенерация, связанная с возрастом.
По патоанатомическим признакам выделены три основные формы дистрофии: доминантные друзы мембраны Бруха, неэкссудативная и экссудативная формы.
К клиническим симптомам относятся постепенное снижение центрального зрения, метаморфопсии, центральная скотома. Друзы являются ранним клиническим проявлением заболевания. Нарушения зрительных функций появляются с развитием макулярной дистрофии. Острота зрения коррелирует с изменениями локальной ЭРГ, в то время как общая ЭРГ остается нормальной (рис. 15.12).
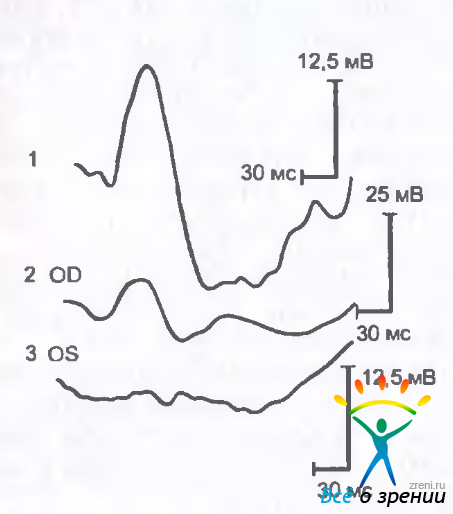
Рис. 15.12. Локальная ЭРГ при разных стадиях макулярной дегенерации, связанной с возрастом. 1 — нормальная, 2 — субнормальная. 3 — нерегистрируемая.
Наиболее часто наблюдается сухая, или атрофическая, форма, которая характеризуется атрофией пигментного эпителия. Реже отмечается экссудативная, "влажная", форма, для которой характерно быстрое ухудшение остроты зрения, связанное с развитием неоваскулярных мембран, фиброваскулярных рубцов и кровоизлияний в сетчатку и стекловидное тело. Отслойка пигментного эпителия часто сочетается с неоваскулярной мембраной и является признаком экссудативной формы возрастной центральной дистрофии сетчатки.
Доминантные друзы мембраны Бруха — двустороннее заболевание с аутосомно-доминантным типом исследования, протекающее бессимптомно. Друзы располагаются в макулярной области перипапиллярно, редко — на периферии глазного дна. Они имеют разные форму, размер и цвет (от желтого до белого), могут быть окружены пигментом.
На ФАГ отмечаются типичные множественные фокальные области ограниченной мелкоточечной поздней гиперфлюоресценции. До сих пор остается невыясненным вопрос, всегда ли друзы предшествуют возрастной, макулярной дистрофии или могут являться самостоятельным заболеванием.
При неэкссудативной форме заболевания обнаруживают друзы в макулярной области и различные проявления патологии пигментного эпителия сетчатки.
Географическая атрофия пигментного эпителия представлена отдельными большими депигментированными зонами, через которые видны крупные хориоидальные сосуды, образующие подковообразное кольцо вокруг фовеальной области, где до последней стадии сохраняется ксантофильный пигмент. Риск образования неоваскулярной мембраны невелик. Географическая атрофия может развиваться на фоне средних и больших друз с нечеткими границами, исчезающего, разрушающегося или отслоенного пигментного эпителия сетчатки; отмечается минерализация друз, которые в этом случае напоминают блестящие ярко-желтые включения.
Негеографическая атрофия не имеет четких границ, выглядит как мелкоточечная гипопигментация в сочетании с гиперпигментацией пигментного эпителия.
Очаговая гиперпигментация может быть самостоятельной патологией или сочетаться с друзами либо прилегающими областями атрофии пигментного эпителия и его отслойки при неоваскуляризации хориоидеи (образование неоваскулярной мембраны). Разрывы пигментного эпителия являются осложнением отслойки сетчатки и обусловлены возникающим натяжением тканей.
Экссудативная форма макулярной дегенерации, связанной с возрастом, проявляется экссудативной отслойкой сенсорной сетчатки с субретинальными геморрагиями и экссудацией липидов, грязно-серым или желтым отеком макулы (цистоидный макулярный отек), образованием складок хориоидеи, отслойкой пигментного эпителия, субретинальным фиброзом. Субретинальный экссудат обычно непрозрачный из-за высокой концентрации протеинов, липидов, продуктов крови, наличия фибрина. Утолщение и серозная отслойка пигментного эпителия сетчатки возникают вследствие образования неоваскуляризации под пигментным эпителием.
Хориоидальная неоваскуляризации представляет собой прорастание кровеносных сосудов через мембрану Бруха в пигментный эпителий. Под нейро- и пигментный эпителий просачиваются кровь, липиды и плазма. Они стимулируют фиброз, который разрушает пигментный эпителий и наружные слои сетчатки. Предполагают, что перекисное окисление липидов в пигментном эпителии способно индуцировать внутриглазную неоваскуляризацию вследствие освобождения цитокинов и других факторов роста. ФАГ помогает в диагностике хориоидальной неоваскуляризации.
Лечение направлено на замедление патологического процесса. С этой целью прежде всего используют антиоксиданты. Клинические наблюдения показывают, что применение ?- и ?-каротина, криптоксантина, селена и других препаратов, обладающих антиоксидантны-ми свойствами, замедляет течение центральной хориоретинальной дистрофии. Аналогично действие витаминов Е и С. Поскольку цинк, вовлеченный во многие энзиматические процессы метаболизма протеинов и нуклеиновых кислот, содержится в большом количестве в комплексе пигментный эпителий сетчатки — xориоидея, предполагают, что прием цинксодержащих препаратов также должен замедлять развитие макулярной дистрофии. Рекомендуется диета, богатая фруктами и овощами.
Для предотвращения деструктивных процессов в сетчатке необходимо применение оптических и фармакологических средств защиты и профилактики, поэтому больным с макулярной дистрофией, связанной с возрастом, рекомендуется назначать, помимо антиоксидантов, сосудистые препараты и липотропные средства, ношение светозащитных очков.
При экссудативной форме заболевания проводят лазерную фотокоагуляцию, ориентируясь на результаты ФАГ-диагностики.
С помощью хирургических методов удаляют хориоидальные неоваскулярные мембраны и субретинальные кровоизлияния. В настоящее время разрабатывают операции по трансплантации пигментного эпителия и фоторецепторного слоя сетчатки. У пациентов с субфовеальной хориоидальной неоваскуляризацией получены положительные результаты фотодинамической терапии. Заболевание хроническое, протекает медленно и приводит к снижению остроты зрения.
Продолжение в следующей статье: Сетчатка ? Часть 3
----
Статья из книги: Глазные болезни | Копаева В.Г.

Комментариев 0