Врожденная глаукома


Содержание:
Описание
Врожденную глаукому классифицируют на простую (первичную), сочетанную и вторичную. Различают инфантильную (до 3 лет) и ювенильную врожденную глаукому. Повышение ВГД у больных с ПВГ и СВГ связано с аномалиями развития дренажной области глаза.↑ Развитие дренажной области глаза
На 6-й неделе беременности по краю оптического бокала возникает недифференцированная клеточная масса, происходящая, по-видимому, из неврального гребешка. Затем недифференцированные клетки распространяются между поверхностной эктодермой и хрусталиком, формируя три слоя:
- роговичного эндотелия;
- стромы роговицы;
- радужки и пупиллярной мембраны [Johnstone М. et al., 1979].
Иридопупиллярная пластинка формируется из мезенхимальной ткани на 2-м месяце развития плода (длина плода 18 мм). Она васкуляризируется сначала из гиалоидной системы, а затем из периферического кольцевого сосуда. Гиалоидные сосуды атрофируются, начиная с 7-го месяца (200 мм), что приводит к атрофии пупиллярной мембраны. Из мезенхимальной ткани дифференцируются также роговичный эндотелий и строма (20 мм). Передняя камера появляется в конце 4-го месяца (110 мм) в виде узкой щели. Изнутри она покрыта непрерывным слоем эндотелия, образуя таким образом замкнутую полость [Hansson Н., Jerndal Т., 1971].
Механизмы развития передней камеры и ее угла не вполне ясны. Важную роль играет быстрый и неравномерный рост ограничивающих ее мезенхимальных слоев. Этот процесс обусловливает возникновение передней камеры, ее углубление, прогрессирующее смещение УПК кзади, растяжение и разрежение тканевых структур [Anderson D., 1981]. Определенное влияние, по-видимому, оказывают также атрофия и резорбция мезенхимальной ткани в области зрачка и УПК [Mann I., 1964].
Задержка в развитии и дифференциации УПК и дренажной системы глаза проявляется в переднем прикреплении корня радужки, чрезмерном развитии гребенчатой связки, заднем положении шлеммова канала, частичном сохранении мезодермальной ткани и эндотелиальной мембраны в бухте угла и на внутренней поверхности трабекул.
Первые признаки склерального синуса в виде сплетения венозных канальцев появляются в конце 3-го месяца (60 мм). Канальцы постепенно сливаются, образуя к 6-му месяцу (150 мм) круговой широкий сосуд. Склеральная шпора начинает формироваться в начале 5-го месяца (110 мм) между синусом и цилиарным телом [Barber A. N., 1955]. К этому времени в цилиарном теле образуются волокна меридиональной цилиарной мышцы, которые достигают кпереди зачатка увеальной трабекулы.
На стадии 150 мм мезенхимальная ткань в УПК дифференцируется в корнеосклеральную и увеальную трабекулы. Увеальная трабекула переходит на цилиарное тело и корень радужки. Трабекулы покрыты изнутри непрерывным слоем эндотелия (мембрана Баркана).
В дальнейшем передняя камера углубляется, УПК распространяется кзади. В том же направлении смещаются корень радужки и цилиарное теле. Так, к 6-му месяцу вершина УПК находится на уровне начала трабекулы, к 7 мес — на уровне середины трабекулы и к моменту рождения доходит до склеральной шпоры. Одновременно происходят атрофия и реорганизация мезенхимальной ткани в УПК и эндотелиальной мембраны Баркана.
Задержка в развитии и дифференциации УПК встречается довольно часто. Она проявляется в чрезмерном развитии гребенчатой связки и отростков радужки, небольшой глубине передней камеры, переднем прикреплении радужки, заднем положении склерального синуса, частичном сохранении мезенхимальной ткани в рецессе УПК.
↑ Гониодисгенез
УПК у детей 1-го года жизни имеет некоторые особенности. Корень радужки выглядит более плоским и тонким, чем у взрослых, увеальная трабекула имеет вид гладкой гомогенной мембраны, идущей от периферии радужки к кольцу Швальбе, в нише УПК и зоне трабекулы иногда видна сероватая вуаль.
Задержка в развитии и дифференциации УПК у 7—8-месячного плода получила название гониодисгенеза. Наиболее выраженным признаком дисгенеза УПК служит переднее прикрепление периферии радужки (рис. 37).

Рис. 37. Гониодисгенез, переднее прикрепление радужки в УПК.
При этом отсутствует вершина угла и кажется, что корень радужки начинается на уровне склеральной шпоры трабекулы или даже кольца Швальбе. В других случаях ниша УПК заполнена тяжами или слоями увеальной ткани. Они отходят от корня радужки, огибают вершину угла и переходят на склеральную шпору и трабекулу. Из этой ткани формируются увеальная трабекула, гребенчатая связка и отростки радужки (рис. 38).
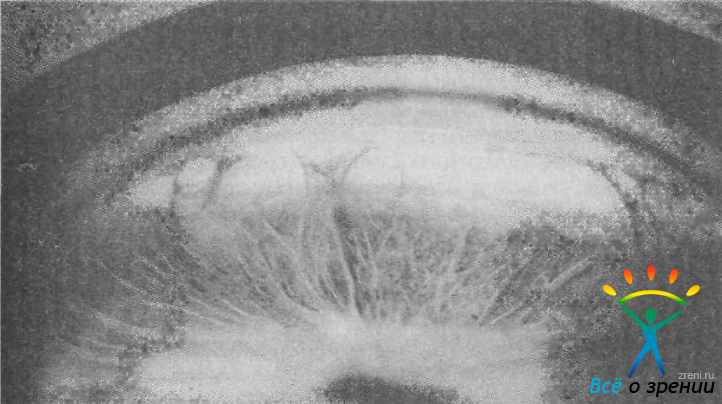
Рис. 38. Увеальная ткань и отростки радужки в УПК у больного с врожденной глаукомой (синдром Ригера).
У детей раннего возраста остатки эмбриональной увеальной ткани видны особенно отчетливо. Они могут быть покрыты изнутри непрерывной эндотелиальной мембраной (мембрана Баркана), закрывающей доступ водянистой влаги к трабекулярному аппарату. Т. Jerndal и соавт. (1978) обнаруживали такую же мембрану во многих глазах с глаукомой у взрослых. Избыток увеальной ткани в УПК часто сочетается с гипоплазией стромы корня радужки. Овальные участки истонченной стромы иногда окаймляются тонкими сосудами. Аномальные циркулярные и радиальные сосуды могут быть обнаружены в нише УПК и корне радужки.
Э. Г. Сидоров и М. Г. Мирзоянц (1988) различают три степени гониодисгенеза. При гониодисгенезе I степени УПК гониоскопически почти не отличается от нормального, отмечается только нежная сероватая вуаль в нише угла и трабекулярной зоне. Дисгенез УПК II степени особенно часто встречается при ВГ. При гониоскопии в плоскости радужки можно видеть, что ее корень прикрепляется на Уровне задней трети аномальной трабекулярной зоны. В глазах со светлой радужкой ее корень имеет зазубренный, фестончатый вид, а в УПК видна полупросвечивающая сероватая ткань. В темноокрашенных глазах виден частокол гребенчатой связки, нередко сливающийся в сплошной пласт, который может продолжаться до передней трети трабекулы. При гониодисгенезе III степени радужка прикрепляется к средней или передней трети трабекулы.
↑ Простая врожденная глаукома
Наследственность. Простая врожденная глаукома (ПВГ) — редкое наследственное заболевание, обнаруживаемое с частотой 1:12 500 рождений [Duke-Elder S., 1964]. ПВГ чаще проявляется на 1-м году жизни и в большинстве случаев (80%) имеет двусторонний характер. Мальчики заболевают чаще девочек. Наследственная передача осуществляется или по аутосомно-рецессивному, или по мультифакториальному типу Morin J., Merin S., 1972]. Однако, по мнению Т. Jerndal (1970), наследуется не глаукома, а дисгенез УПК, передающийся по доминантному типу. В зависимости от экспрессивности дисгенеза возникает инфантильная, ювенильная, первичная открытоугольная глаукома или глаза остаются клинически здоровыми в течение всей жизни.
Вопрос об общем генетическом базисе ПВГ и первичной глаукомы у взрослых нуждается в дополнительном изучении. По данным Т. И. Ершевского и Р. П. Шикуновой (1978), такой общий базис существует. Однако J. Morin и S. Merin (1972) установили, что в семьях больных с ПВГ пораженность первичной глаукомой такая же, как и в нормальной популяции. Они указывают также на преобладание отрицательного кортикостероидного теста у детей с врожденной глаукомой в отличие от больных первичной ОУГ. По-видимому, общин генетический базис ПВГ первичная ОУГ имеет только в тех случаях, когда в ее патогенезе значительную роль играет дисгенез УПК. S. Phelps и S. Podos (1974) показали, что НLА-антигены не информативны как генетические маркеры при врожденной глаукоме. Если в семье есть ребенок с ПВГ, то риск рождения второго ребенка с тем же заболеванием составляет 1:20 [Jog М., Rice N,, 1978].
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Патогенез. Патогенез ПВГ связывают с дисгенезом УПК. Основываясь на результатах гониоскопических и гистологических исследований, O. Barkan (1949, 1955) выдвинул теорию, согласно которой в глазах с инфантильной глаукомой существует претрабекулярная мембрана которая блокирует фильтрующую зону УПК. Позднее L. Allen и соавт. (1955) пришли к выводу, что врожденная глаукома является следствием неправильного и неполного расщепления тканей в УПК в процессе эмбриогенеза. E.Maumeneе (1958) выдвинул новую концепцию, согласно которой инфантильная глаукома является следствием прикрепления продольных волокон цилиарной мышцы не к склеральной шпоре, а дальше кпереди — к корнеосклеральной трабекуле.
A. Towara и H. Inomata (1987) описали слой субканаликулярной плотной ткани у больных врожденной глаукомой. Этот слой состоит из клеток с короткими цитоплазматическими отростками и экстрацеллюлярной субстанции. У больных инфантильной глаукомой он был толще, чем в глазах с ювенильной глаукомой. Авторы считают, что описанный ими слой субканаликулярной ткани — следствие неполного развития трабекулярной сети и он может служить причиной возникновения глаукомы в любом возрасте.
Концепцию, предложенную O.Barkan, разделяют I. Worst (1966), а также Т. Jerndal и соавт. (1978), которые в глазах с ПВГ обнаружили более плотную увеальную трабекулу и слабо фенестрированный слой эндотелия на внутренней поверхности трабекулярного переплета. Основываясь на результатах патоморфологических исследований, Э. Г. Сидоров и М. Г. Мирзоянц (1987) указывают на возможную роль в генезе глаукомы неправильно сформированной увеальной трабекулы и всего трабекулярного аппарата, рудиментарного состояния склеральной шпоры и шлеммова канала, чрезмерно заднего положения последнего, вплетения волокон цилиарной мышцы непосредственно в трабекулу. Они так же, как и D. Anderson (1981), не обнаружили мембраны Баркана при патогистологических исследованиях.
Таким образом, по данным разных авторов, наиболее частыми причинами нарушения оттока водянистой влаги из глаза при ПВГ служат сохранение эндотелиальной мембраны Баркана в УПК, остатки увеальной эмбриональной ткани в рецессе УПК и трабекулярной зоне (включая гребенчатую связку и отростки радужки), переднее прикрепление радужки, дефекты в формировании трабекулярного аппарата и шлеммова канала, аномалии в топографии цилиарной мышцы.
Клиника. Клиника инфантильной глаукомы имеет специфические особенности. У ребенка появляются светобоязнь и слезотечение, вызванные растяжением и отеком роговицы. Он не поворачивает голову к свету, а, наоборот, отворачивается от него. В тяжелых случаях возникает блефароспазм. Может выявляться симптом красного глаза. Характерные изменения обнаруживают в роговице, передней камере, УПК, радужке и ДЗН.
Горизонтальный диаметр роговицы у здорового новорожденного равен 10 мм, увеличиваясь до 11,5 мм к 1 году к до 12 мм к 2 годам. У больных инфантильной глаукомой диаметр роговицы уже на 1-м году жизни увеличен до 12 мм и больше, уменьшена толщина роговицы и увеличен радиус ее кривизны. Растяжение роговицы часто сопровождается отеком стромы и эпителия и разрывами десцеметовой оболочки, которые можно обнаружить с помощью лупы или ручной щелевой лампы. В поздних стадиях болезни происходит Рубцевание стромы и возникают стойкие помутнения в роговице.
Для врожденной глаукомы характерны углубление передней камеры, атрофия стромы радужки, обнажение ее радиальных сосудов. Однако следует отметить, что и у здоровых новорожденных строма Радужки бывает слабо развита, особенно в периферической зоне.
Нормальное глазное дно в неонатальном периоде бледное из-за неполного развития пигментного эпителия. Диск зрительного нерва бледнее, чем у взрослого, физиологическая экскавация отсутствует или слабо развита. При врожденной глаукоме экскавация быстро увеличивается в размерах и становится глубокой. Следует отметить, что сначала экскавация ДЗН носит обратимый характер и уменьшается при снижении ВГД. По данным J. Morin и соавт. (1974), увеличение отношения диаметров Э/Д на 0,2 соответствует приросту диаметра роговицы на 0,5 мм. Это позволяет ориентировочно оценить состояние ДЗН без офтальмоскопии.
При измерении с помощью ультразвука длина оси глаза новорожденного варьирует от 17 до 20 мм, достигая 22 мм к концу 1-го года жизни. При глаукоме размеры глазного яблока увеличиваются, иногда весьма значительно, но могут быть и в пределах нормальных значений. Следует отметить, что изменения диаметра роговицы имеют большее значение в диагностике ПВГ и оценке стадии болезни, чем увеличение длины оси глаза.
Данные о величине нормального ВГД у новорожденных и детей 1-го года жизни противоречивы. Это связано с трудностью измерения давления у детей, а также с тем, что оно меняется под воздействием наркотических средств. Э. Г. Сидоров и М. Г. Мирзоянц (1987) установили, что при использовании кеталаровой анестезии верхняя граница нормального ВГД у детей такая же, как и у взрослых. Однако при проведении фторотанового наркоза ВГД снижается на 2—3 мм рт.ст. У детей с врожденной глаукомой часто наблюдаются значительные колебания офтальмотонуса в течение суток от нормальных значений до 40 мм рт.ст. и выше.
В поздней стадии болезни глаз и особенно роговица значительно увеличены, роговичный лимб растянут, плохо контурирован, роговица мутная, нередко проросшая сосудами. Глаз в таком состоянии называют «бычьим» (buphtalm). Перерастяжение и разрыв цинновых связок приводит к иридодонезу и сублюксации хрусталика. В слепом глазу нередко возникают язвы роговицы, гифемы, может произойти перфорация язвы или разрыв истонченных оболочек глазного яблока с исходом во фтизис глаза.
Врожденную инфантильную глаукому нужно дифференцировать от мегалокорнеа, поражений роговицы у детей, травматических разрывов десцеметовой оболочки и врожденного дакриоцистита. Мегалокорнеа — врожденная наследственная аномалия роговицы. В отличие от глаукомы при мегалокорнеа роговица прозрачная, роговичный лимб четко очерчен, не растянут, роговицы обоих глаз одинаковые по размеру, толщине и кривизне. Однако следует иметь в виду, что в редких случаях возможно сочетание двух заболеваний — мегалокорнеа и врожденной глаукомы.
Помутнение роговицы у детей раннего возраста может быть при цистинозе, мукополисахаридозе, врожденной дистрофии роговицк, кератитах. Однако при этих заболеваниях отсутствуют другие симптомы, характерные для врожденной инфантильной глаукомы. Единственным общим симптомом врожденного дакриоцистита и ПВГ является слезотечение. Однако в первом случае отсутствуют светобоязнь и изменения роговицы, а во втором — нет гнойного содержимого в конъюнктивальной полости.
Клиническая симптоматика ювенильной ПВГ существенно отличается от проявлений инфантильной глаукомы. Роговица и глазное яблоко имеют нормальные размеры, отсутствуют светобоязнь, слезотечение и все симптомы, связанные с растяжением и отеком роговицы. Вместе с тем, как и при инфантильной глаукоме, может наблюдаться феномен растяжения склерохориоидального канала зрительного нерва. Общим с инфантильной глаукомой является состояние УПК, характеризующееся его дисгенезом той или иной степени выраженности.
Медикаментозное лечение ПВГ малоэффективно. Предпочтение отдают хирургическому лечению, которое не следует откладывают [Брошевский Т. И., Токарева Б. А., 1971; Ковалевский Е. И., Татаринов С. А., 1982]. Выбор операции зависит от стадии болезни, особенностей строения УПК и опыта хирурга. В ранней стадии болезни чаще проводят гониотомию [Брошевский Т. И., Токарева Б. А., 1971; Краснов М. М., 1980] или трабекулотомию [Сидоров Э. Г., Мирзоянц М. Г., 1987]. В поздних стадиях ПВГ более эффективны фистулизирующие операции и деструктивные вмешательства на цилиарном теле [Ковалевский Е. И., Татаринов С. А., 1982].
Прогноз при своевременном проведении хирургического лечения удовлетворительный. Устойчивой нормализации ВГД удается добиться в 85% случаев. Зрение сохраняется в течение всей жизни у 75% больных, которым операция произведена в начальной стадии болезни, и только у 15—20% поздно оперированных больных.
↑ Сочетанная врожденная глаукома
Сочетанная врожденная глаукома (СВГ) имеет много сходного с ПВГ. В большинстве случаев она также развивается вследствие дисгенеза УПК и имеет две формы: инфантильную (у детей до 3 лет) к ювенильную (старше 3 лет). Особенно часто врожденная глаукома сочетается с аниридией, микрокорнеа, персистирующим первичным стекловидным телом, мезодермальным дисгенезом, факоматозами, синдромами Марфана и Маркезани, хромосомными нарушениями, а также с синдромами, вызванными внутриутробным поражением вирусом краснухи.
Микрокорнеа. К микрокорнеа относят случаи с горизонтальным диаметром роговицы меньше 10 мм. Небольшие размеры роговицы часто сочетаются с мелкой передней камерой и узким УПК. Глаукома в глазах с микрокорнеа чаще протекает по типу закрытоугольной, однако описаны также случаи открытоугольной врожденной глаукомы.
Персистирующее гиперпластическое первичное стекловидное тело. В типичном случае в глазу с микрофтальмом позади хрусталика видны белые массы гиперплазированного первичного стекловидного тела. Сохраняются также остатки гиалоидной артериальной системы. Хрусталик набухает и мутнеет, возникают зрачковый блок и закрытоугольная глаукома. В других случаях глаукома носит вторичный характер, развиваясь после кровоизлияний в стекловидное тело. Белые массы за хрусталиком могут послужить причиной установления ошибочного диагноза ретинобластомы.
Аниридия и глаукома. По данным M. Shaw и соавт. (1960), врожденная аниридия встречается примерно в двух случаях на 100 000 рождений. Она может быть одиночным дефектом или сочетаться с другими врожденными аномалиями. Наследственная передача в большинстве случаев осуществляется по аутосомно-доминантному типу, но возможны также и аутосомно-рецессивная передача и возникновение дефекта радужки за счет спонтанных мутаций.
В типичных случаях радужка отсутствует почти полностью, за исключением небольшой периферической «культи». Однако у некоторых больных дефект радужки менее выражен и она в той или иной степени сохранена. Из других поражений глаз описаны васкуляризация периферии роговицы, нарушение ее эпителия, врожденные помутнения в хрусталике, Колобома хориоидеи, макулярная гипоплазия, частичный птоз, нистагм. В спорадических случаях аниридия сочетается с опухолью Вилмса (смешанная опухоль почки), которая может метастазировать в орбиту.
Глаукома развивается у 50—75% больных с аниридией (чаще в возрасте 5—15 лет) и протекает по ювенильному типу. Патофизиологические механизмы глаукомы при аниридии связаны как с дисгенезом угла передней камеры и дренажной системы глаза, так и особенно с последующими, вторичными, изменениями в этих структурах. Вторичные изменения заключаются в васкуляризации «культи» радужки, ее прогрессирующем сращении с трабекулярной стенкой УПК и его облитерации.
Лечение начинают с назначения гипотензивных препаратов. В случае отсутствия достаточного эффекта рекомендуется хирургическое лечение. Выбор операции зависит от конкретного случая. Из фистулизирующих операций предпочтительнее фильтрующая иридоциклоретракция. В части случаев достаточный гипотензивный эффект достигается с помощью циклокриокоагуляции.
Мезодермальный дисгенез переднего отдела глаза. Проявления мезодермального дисгенеза переднего отдела глаза многообразны как по клинической картине, так и по интенсивности. Ниже будут рассмотрены только те из них, которые нередко ассоциируются с вторичной ювенильной или юношеской глаукомой. Дисгенез переднего отдела глаза принято делить на периферический и центральный.
Периферический мезодермальный дисгенез. В эту группу входят задний эмбриотоксон, аномалия Аксенфельда и синдром Ригера. Задним эмбриотоксоном Т. Axenfeld (1920) назвал выраженное проминирование и смещение кпереди переднего пограничного кольца Швальбе. Эта аномалия встречается довольно часто и сама по себе не служит причиной каких-либо глазных заболеваний. Вместе с тем задний эмбриотоксон нередко сочетается с более глубокими проявлениями мезодермального дисгенеза. Диагностика заднего эмбриотоксона несложна. При биомикроскопии на периферии роговицы видна белая полоска, а при гониоскопии — выступающее кзади кольцо Швальбе. Аномалию Axenfeld в настоящее время рассматривают как «мягкий» вариант более тяжелого синдрома, описанного Н. Rieger(1935).
Синдром Ригера — наследственное двустороннее заболевание с аутосомно-доминантным типом передачи. Выраженность синдрома у членов одной семьи значительно варьирует. Наиболее характерным глазным признаком болезни является синдром Аксенфельда, т. е. задний эмбриотоксон и отростки или тяжи радужки, идущие от ее периферии, а иногда и от зрачковой зоны к кольцу Швальбе (см. рис. 38). Одновременно отмечаются признаки гипоплазии стромы радужки, сочетающиеся со зрачковыми дефектами (дислокация зрачка, нарушение его формы, выворот пигментного листка). В более тяжелых случаях гипоплазия захватывает и пигментный слой, в результате чего в радужке образуются отверстия (рис. 39).
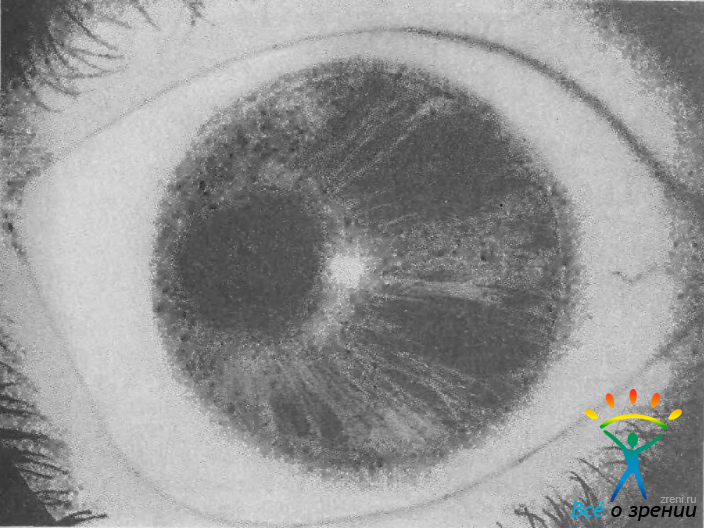
Рис. 39. Гипоплазия радужки, деформация и дислокация зрачка у больного с синдромом Ригера и врожденной глаукомой.
Изменения в радужке обычно стационарные, но иногда прогрессируют, вероятно, вследствие недостаточного развития сосудов и ишемии. У некоторых больных отмечаются изменения величины и формы роговицы (мегало- или микрокорнеа, вертикально-овальная роговина), поражения хориоидеи, сетчатки, катаракта, косоглазие.
Глазные изменения часто сочетаются с аномалиями зубов и лицевого черепа. У больных с синдромом Ригера нередко уменьшен!, количество и размеры зубов, увеличены промежутки между ними, отмечаются гипоплазм верхней челюсти, расширенная плоская переносица, выпяченная нижняя губа.
Изменения в переднем отделе глаза примерно у половины больных ведет к развитию глаукомы, которая обычно проявляется в детском или юношеском возрасте. Механизм повышения внутри глазного давления связан не только с отростками радужки. Получены данные, свидетельствующие о том, что главную роль играют дефекты в развитии трабекулы и склерального синуса. Имеет значение и обычное для синдрома Ригера переднее прикрепление радужки к трабекулярной зоне.
Синдром Ригера следует дифференцировать прежде всего от мезодермальной дистрофии радужки. Клинические проявления этих заболеваний очень сходны. Можно указать на следующие различия. Для глаукомы, вызванной синдромом Ригера, характерны положительный семейный анамнез, начало в детском возрасте (часто, но не всегда), поражение обоих глаз, отсутствие отека роговицы, изменения зубов и лицевого черепа. У больных с мезодермальной дистрофией радужки заболевание начинается позднее, часто в среднем возрасте, семейный анамнез редко бывает положительным, поражение может быть односторонним, возможен отек роговицы из-за дефекта роговичного эндотелия. Значительно легче дифференцировать синдром Ригера от иридошизиса, корэктопии, аниридии и врожденной гипоплазии радужки вследствие заметных различий в клинической картине этих заболеваний.
Лечение глаукомы, ассоциирующейся с синдромом Ригера, заключается в применении лекарственных препаратов, снижающих продукцию водянистей влаги (тимолол, клофелик), в легких случаях и в выполнении оперативного вмешательства — в более тяжелых.
Центральный мезодермальный дисгенез. К этой группе пороков развития относят задний кератоконус, роговичную аномалию Питерса, а также врожденные бельма и стафиломы роговицы. Можно предположить, что перечисленные выше дефекты развития представляют собой одну и ту же аномалию, но разной степени выраженности. Для нее характерно поражение задних слоев роговицы в ее центральном отделе.
При заднем кератоконусе отмечается увеличение кривизны задней поверхности роговицы в ее центральном сщеле. Аномалия Питерса [Peters А., 1906] характеризуется центральным помутнением роговицы, а также дефектом десцеметовой мембраны и эндотелия в зоне помутнения. При этом задние слои роговицы сращены с центральными отделами радужки или хрусталиком. В последнем случае изменения в роговице сочетаются с катарактой. Полагают, что аномалия Питерса — наследственное заболевание с аутосомно-рецессивным типом передачи. При аномалии Питерса обычно поражаются оба глаза, иногда она сочетается с микрофтьльмом, голубой склерой и синдромом Ригера. Аномалия Питерса часто осложняется глаукомой, которая развивается сразу же после рождения ребенка.
Врожденные бельма роговицы в наиболее тяжелых случаях сочетаются со стафиломой. При этом роговица истончена, васкуляризована и сращена с радужкой, а внутриглазное давление нередко повышено.
Аномалию Питерса дифференцируют от ПВГ, помутнений роговицы, вызванных родовой травмой, врожденной роговичной дистрофии, мукополисахаридоза.
Возможно только хирургическое лечение глаукомы, ассоциирующейся с центральным дисгенезом переднего отдела глаза (трабекулэктомия, фильтрующая иридоциклоретракция, криоциклокоагуляция). После нормализации ВГД показана сквозная кератопластика.
Синдром Франк-Каменецкого. Для этого синдрома характерно сочетание гипоплазии стромы радужки с врожденной глаукомой. Заболевают мальчики. Болезнь передается по рецессивному, сцепленному с полом типу (рис. 40).
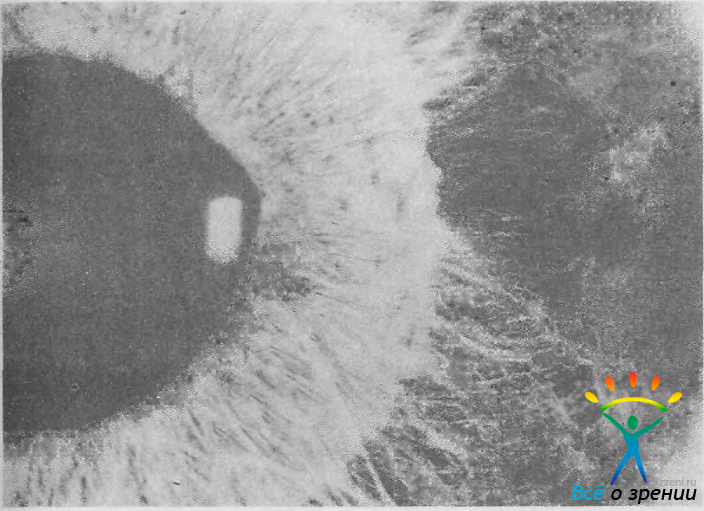
Рис. 40. Характерная картина двухцветной радужки и гипоплазии ее стромы у больного с синдромом Франк-Каменецкого.
Наиболее выраженный синдром — двухцветная радужка: светлая зрачковая зона сочетается с более темной, коричневатой периферией. Темный цвет цилиарной зоны обусловлен гипоплазией стромы радужки и просвечиванием пигментного листка. У части больных встречаются зрачковые аномалии и сквозные отверстия в радужке.
Склерокорнеа. Склерокорнеа — врожденное поражение роговицы, в которую врастает васкуляризованная склеральная ткань. Помутнение захватывает или периферию, или всю роговицу. Склерокорнеа может сочетаться с другими общими и глазными врожденными изменениями, включая глаукому. Причинами повышения давления в глазу служат или облитерация УПК за счет иридокорнеальные сращений, или дисгенез УПК и дренажной системы глаза. Для восстановления зрения рекомендуется кератопластика; при сочетании склерокорнеа с глаукомой прогноз плохой. Возможно только хирургическое лечение глаукомы.
Синдром Марфана (арахнодактилия). Синдром Марфана представляет собой наследственную системную гипопластическую мезенхимальную дистрофию. Заболевание передается по аутосомно-доминантиому типу с высокой пенетрантностью, Наиболее выражены изменения скелета: арахнодактилия, долихоцефалия, длинные, тонкие конечности, кифосколиоз, ослабленные связки и суставы. Характерны также кардиоваскулярные нарушения, особенно изменения в аорте.
Наиболее частые глазные изменения — увеличение размеров глазного яблока, истончение оболочек и диск локация хрусталика (ectopia lentis), которая отмечается у 60—80 % больных. Хрусталик, часто уменьшенный в размерах и сферической формы, смещается, как правило, кверху. У части больных развивается инфантильная или ювенильная глаукома. В таких случаях при гистологическом исследовании обнаруживают элементы дисгенеза УПК: переднее прикрепление меридиональных волокон цилиарной мышцы, слабое развитие склеральной шпоры, утолщение трабекулярной сети, иногда неполное развитие склерального синуса. Лечение глаукомы при синдроме Марфана может быть медикаментозным или хирургическим в зависимости от конкретного случая.
Гомоцистинурия. Внешние общие проявления болезни такие же, как при синдроме Марфана. В отличие от последнего гомоцистинурия передается по аутосомно-рецессивному типу и часто сопровождается задержкой умственного развития. Нарушение гомоцистеинового метаболизма является следствием энзимного дефекта. Дислокация хрусталика и глаукома наблюдаются чаше, чем при синдроме Марфана. Заболевание может осложняться отслойкой сетчатки.
Синдром Маркезани (сферофакия—брахиморфия). Синдром Маркезани представляет собой наследственное системное заболевание гиперпластического типа, которое может передаваться по доминантному или рецессивному типу. Больные — брахицефалы, невысокого роста с короткими широкими конечностями и пальцами, хорошо развитой подкожной тканью и мускулатурой. Глазные изменения включают микросферофакию, хрусталиковую миопию, иногда дислокацию хрусталика (чаще книзу). Глаукома развивается не часто, она может быть как открыто-, так и закрытоугольной. В первом случае повышение ВГД бывает связано с дисгенезом УПК, во втором — с блоком зрачка сферическим хрусталиком.
Окулоцереброренальный синдром. Синдром описан C. Lowe, М. Теrrу и E. Maclochlan (1952). Из основных симптомов следует отметить системный ацидоз, увеличенную органическую ацидурию, кетонурию, глюкозурию, альбуминурию, аминоацидурию, мышечные, скелетные и нервно-психические нарушения. Глаукома развивается более чем у половины больных и протекает по типу инфантильной. Характерны также врожденные катаракты и помутнения роговицы. Лечение глазных проявлений болезни заключается в экстракции катаракты и хирургическом лечении глаукомы (трабекулотомия или трабекулэктомия).
Другие синдромы. Врожденная глаукома в редких случаях может сочетаться и с другими нарушениями, включая синдромы Дауна, Робина, Турнера Стиклера, ретиноцеребральный ангиоматоз, оку-лодермомеланоцитоз, хромосомное синдромы (трисомия 13—15, 17—18). Клиническое течение глаукомы в таких случаях аналогично первичной инфантильной глаукоме.
Энцефалотригеминальный ангиоматоз (синдром Стюржа-Вебера). Синдром Стюржа-Вебера относят к факоматозам — наследственным поражениям различных органов, характеризующихся развитием опухолевидных образований, гиперплазией тканей, возникающих из обычных тканевых клеток (гамартомы), или развитием истинных опухолей из недифференцированных эмбриональных или измененных взрослых клеток. Глаукома как редкое осложнение может возникать и при таких факоматозах, как нейрофиброматоз Рекликгхаузена, окулодермалышй меланоцитоз, ангиоматоз сетчатки (болезнь Гигшеля—Линдау), туберозный склероз, диффузный врожденный гемангкоматоз. Однако как отдельную клиническую форму можно выделить только глаукому, ассоциированную с синдромом Стюржа—Вебера.
Синдром включает в себя ангиоматозное поражение лица, мягкой мозговой оболочки и глаз. У части больных поражаются только лицо и глаза или лицо и мягкая мозговая оболочка. Ангиоматоз может быть более распространенным: ангиомы образуются в полости рта, носа и в других органах.
Наиболее постоянным и выраженным симптомом является кожная ангиома на лице. Ангиома насыщенно-красного цвета локализуется в зоне разветвления первой: и второй ветвей тройничного нерва, особенно часто захватывая супраорбитальную область. Обычно, но не всегда, поражается только одна сторона лица.
Ангиоматозное поражение мягких мозговых оболочек чаще локализуется в затылочной области, где происходит кальцификация артерии и облитерация ген. Вследствие этого у больных возникают различные неврологические симптомы.
В глазу гемангиому обнаруживают в конъюнктиве, эписклере и хориоидее. Реже поражаются другие отделы сосудистой оболочки, иногда — ткани орбиты. Ангиома хориоидеи относится к кавернозному типу и имеет вид несколько приподнятого желтовато-оранжевого образования. Размеры ее индивидуально варьируют, иногда она захватывает почти всю хориоидеи).
К диагностике глазных поражений при синдроме Стюржа—Вебера имеет значение «правило верхнего века»: если в процесс вовлечено верхнее веко, следовательно, есть и поражение глаза, и, наоборот, отсутствие ангиомы на верхнем веке свидетельствуем об отсутствии поражения глаза. Однако встречаются исключения из этого правила.
По данным G. Alexander и A. Norman (1960), глаукома развивается у каждого 3-го больного с синдромом Стюржа—Вебера. При этом у 60% больных она «рожденная и у 40% возникает в более позднем возрасте. Врожденная глаукома часто заканчивается развитием буфтальма и слепоты. Более поздняя глаукома протекает по типу ОУГ или хронической ЗУГ. Обычно поражается один глаз, реже заболевание бывает двусторонним.
Существуют различные точки зрения на механизмы повышения ВГД при синдроме Стюржз—Вебера. Решающую роль, по-видимому, играют дисгенез УПК, дефекты развития дрензжной системы глаза и повышение эписклерального венозного давления [Weiss D. I., 1973]. Последний фактор связан с эписклеральными гемангиомами и артериовенозными шунтами.
Лечение глаукомы у больных с синдромом Стюржа—Вебера — трудная проблема. Только в легких случаях достаточно назначения гипотензивных средств. Из оперативных вмешательств чаще используют трабекулэктомию. Следует иметь в виду, что резкое снижение ВГД может привести к тяжелым осложнениям. Обильная транссудация жидкости из хориоидальной ангиомы вызывает смещение содержимого глаза кпереди, вплоть до выпадения в рану стекловидного тела. Значительно повышен также риск возникновения экспульсивного кровотечения. Из других осложнений следует отметить упорное кровотечение из эписклеральных сосудов и перерезанных концов склерального синуса с образованием рецидивирующих гифем. Для предупреждения этих осложнений следует максимально снизить ВГД перед операцией, сделать профилактическую заднюю склерэктомию (два отверстия в разных сегментах), снизить АКД. Резекцию глубокой лимбальной пластинки следует производить кпереди от склерального синуса, избегая его повреждения. Л. В. Вязигина и Ю. Е. Батманов (1985) предложили выключать участок шлеммова какала в зоне планируемой операции между устьями крупных венозных коллекторов с помощью аргонового лазера. Эта манипуляция позволяет уменьшить опасность кровотечения из концов перерезанного канала во время и после операции.
Нейрофиброматоз. Нейрофиброматоз относят к факоматозам. Он представляет собой нейроэктодермальную дисплазию, характеризующуюся пролиферацией периферических нервных элементов с образованием опухолеподобных структур. Заболевание передается по аутосомно-доминантному типу. Основные поражения локализуются в коже, периферической и центральной нервной системах
В глазной практике приходится иметь дело с поражением век, конъюнктивы, орбиты, роговицы, сосудистой оболочки, сетчатки, зрительного нерва. Особенно часто поражается верхнее веко, где образуется плексиформная фиброма, нередко распространяющаяся и на височную область. В процесс обычно вовлекается одна сторона, реже бывают двусторонние изменения. Нейрофиброматозные узелки или диффузная инфильтрация может возникать на конъюнктиве, эписклере, роговице и радужке. Иногда отмечается значительное утолщение хориоидеи и цилиарного тела за счет пролиферации ткани, В зрительном нерве описаны менингиомы к глиомы, в орбите — нейрофибромы.
Глаукома развивается редко, часто сочетается с поражением верхнего века и обычно (но не всегда) бывает односторонней. Причиной повышения давления служат дисгенез УПК, аномалии развития склерального синуса или претрабекулярная блокада нейрофиброматозной тканью. В некоторых случаях развивается закрытоугольная глаукома, вызванная смещением кпереди иридохрусталиковой диафрагмы из-за утолщения хориоидеи и цилиарного тела.
Медикаментозное лечение глаукомы, ассоциирующейся с нейрофиброматозом, бывает успешным только в редких случаях. Выбор метода хирургического лечения зависит от опыта офтальмохирурга и особенностей течения заболевания в конкретном случае. Чаще производят трабекулотомию или трабекулэктомию.
Краснуха. Разнообразные врожденные дефекты обнаруживают у новорожденных, матери которых заболели краснухой в первом триместре беременности. У них отмечаются задержка общего развития, глухота, сердечные расстройства и глазные поражения. Последние включают (в порядке частоты) ретинопатию, косоглазие, катаракту, нистагм, микрофтальм, микрокорнеа, атрофию зрительного нерва, помутнение роговицы, глаукому, дефекты век и атрофию радужки [Wilff S., 1972).
A. Kolker и J. Hetherington (1976) считают, что глаукома развивается у 10—25% детей с доказанным синдромом краснухи. Однако S. Wolff (1972) при обследовании 730 детей с врожденной краснухой обнаружил глаукому только у 22 (3%). Следует иметь в виду, что повышение ВГД у детей с врожденной краснухой нередко бывает временным и связано с внутриглазным воспалительным процессом. Истинная инфантильная глаукома возникает в тех глазах, где произошла задержка развития иридокорнеального угла, имевшая такие же последствия, как при первичной врожденной глаукоме. Понятно, что хирургическое лечение глаукомы показано только в тех случаях, когда точно установлен диагноз инфантильной глаукомы.
↑ Вторичная инфантильная глаукома
Наиболее частыми причинами вторичной инфантильной глаукомы являются
- ретинобластома,
- ювенильная ксантогранулема,
- ретролентальная фиброплазия,
- травмы и воспаления, локализующиеся в заднем отделе глаза.
Ювенильная ксантогранулема представляет собой доброкачественное ксантоматозное поражение кожи. Заболевание возникает в раннем детстве и характеризуется появлением множественных желтовато-оранжевых узелков на коже, которые затем спонтанно исчезают. Глазные поражения заключаются в желтоватого цвета сосудистом утолщении радужки. Глаукома является следствием внутриглазных кровоизлияний. Для лечения используют общие и субконъюнктивальные инъекции стероидов и глазные гипотензивные препараты.
Глаукома при ретролентальной гипоплазии вызвана смещением кпереди иридохрусталиковой диафрагмы, поэтому она протекает по типу ЗУГ.
Механизм возникновения вторичной глаукомы при травмах глаза и внутриглазных воспалительных процессах такой же, как при аналогичных поражениях у взрослых. Основное отличие в клинической картине и течении болезни заключается в тенденции к увеличению размеров роговицы и глаза у детей в возрасте до 3 лет.
----
Статья из книги: Глаукома. Нестеров А.П.

Комментариев 0