Глазная генетика и эмбриология

Содержание:
Описание

↑ Краткие сведения об эмбриогенезе глаза человека
Зачатки глаза у зародыша человека появляются очень рано. На второй неделе внутриутробного развития на дорсальной поверхности медуллярной пластинки появляются глазные ямки. В конце 3-й недели развития при замыкании мозговой трубки из них образуются первичные глазные пузыри, которые перемещаются и принимают боковое положение. В результате более быстрого роста задних и боковых частей первичного глазного пузыря происходит образование вторичного пузыря, состоящего из двух слоев (глазного бокала). Формирующуюся зародышевую щель заполняет прилежащая мезодерма, которая образует первичное мезодермальное СТ и сосудистую сеть хориоидеи. Тогда же, к концу 4-й недели развития эмбриона, из поверхностной эктодермы образуется зачаток хрусталика (рис. 23-1).
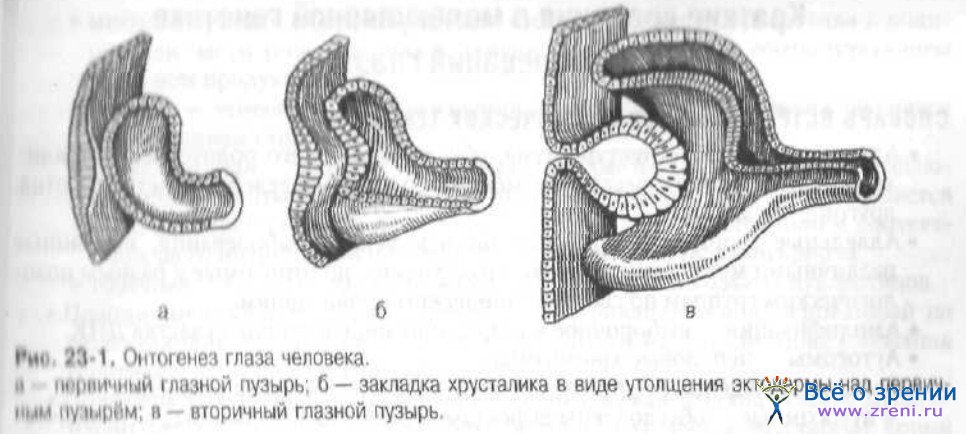
Капсула хрусталика возникает на 5-й неделе эмбрионального развития. На 8-й неделе развития в период образования первичного ядра хрусталика начинают формироваться швы хрусталика.
На 5-6-й неделе происходит закрытие зародышевой щели. Сохранившаяся над хрусталиковым пузырьком эктодерма в дальнейшем дифференцируется в передний эпителий роговицы. Вокруг хрусталикового пузырька формируется сосудистая сумка. Первичное СТ также пронизано большим количеством сосудов. Начинает дифференцироваться ножка глазного бокала, образуется артерия стекловидного тела.
Наружный листок бокала в дальнейшем преобразуется в пигментный слой сетчатки, внутренний даёт начало собственно сетчатке. Прорастая впереди хрусталика, края глазного бокала образуют радужную и ресничную части сетчатки. На 7-й неделе развития нервные волокна входят в канал зрительного нерва. Происходят закладка и развитие век и поперечнополосатых мышц глаза. Из мезенхимы, состоящей из клеток нервного гребешка и краниальной мезодермы, на 8-й неделе происходит закладка склеры. Формируются зрительный нерв, зрительный тракт и частичный перекрест волокон в хиазме. В мезенхиме, которая находится между эктодермой и хрусталиком, появляется щель — передняя камера. Из мезенхимы в дальнейшем формируются строма роговицы, эндотелий, трабекулярная сеть. На 10-й неделе развития происходит дифференцировка нейроэпителиальных клеток на палочки и колбочки, формируются цилиарное тело, его отростки и мышца (рис. 23-2).
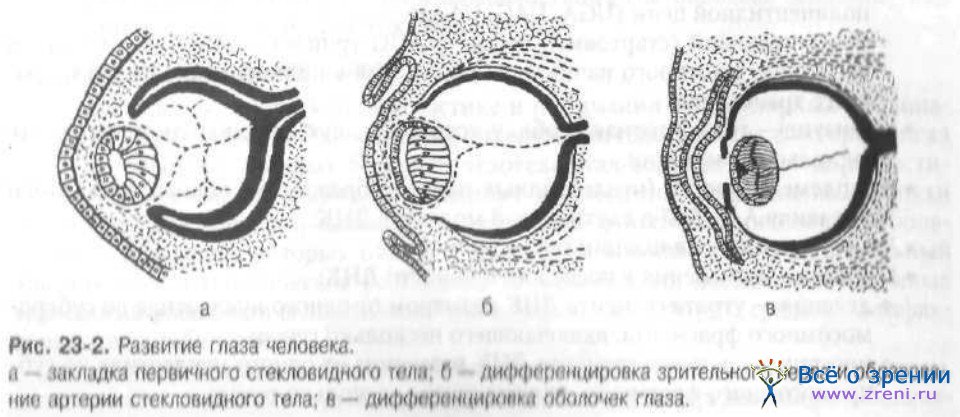
После 12-й недели происходят тонкая дифференцировка всех тканей и формирование функциональных систем. Слёзная железа формируется на 3-м месяце внутриутробного развития, а на 5-м месяце слёзный канал открывается в носовую полость. На 7-м месяце исчезают мембрана, закрывающая зрачок, артерия стекловидного тела, а также спайка между верхним и нижним веком. На 8-м месяце происходит развитие решётчатой пластинки склеры, исчезает сосудистая сумка хрусталика. На 9-м месяце происходит миелинизация волокон хиазмы и зрительного нерва. Полностью исчезают сосуды стекловидного тела, и оно приобретает прозрачность. Завершение некоторых из этих процессов может происходить в течение первых недель после рождения.
В регулировке сложных механизмов пролиферации, дифференцировки и гибели клеток, происходящих в процессе закладки и развития органа зрения, играет важную роль взаимодействие большого количества генов, многие из продуктов которых являются транскрипционными факторами (РАХ6, TGFбетта-семейство, FOXC1, FOXC2, FOXE3, PITX2, PITX3, LMX1B, CYP1B1 и др.). Нарушения в структуре этих генов или их взаимодействия приводят к грубым нарушениям развития. Для гетерозигот по мутациям в генах SOX2, ВМР4, РАХ6 и ряде других характерно наличие выраженного дисгенеза глазных структур. У гомозигот по мутациям в этих генах либо не происходит развития органа зрения, либо они гибнут во внутриутробном периоде вследствие грубых дефектов других органов и тканей (FOXC1, PITX2).
↑ Краткие сведения о молекулярной генетике заболеваний глаза
↑ СЛОВАРЬ ВСТРЕЧАЮЩИХСЯ ГЕНЕТИЧЕСКИХ ТЕРМИНОВ
• Аллель — вариант структуры гена, обусловленный его родительским происхождением (в соматических хромосомах один из аллелей достаётся от отца, другой — от матери).
• Аллельные серии — моногенные наследственные заболевания, вызванные различными мутациями в одном и том же гене, но относимые к разным нозологическим группам по своим клиническим проявлениям.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
• Амплификация — выборочное копирование определённого участка ДНК.
• Аутосомы — неполовые хромосомы.
• Болезни врождённые — присутствуют у ребёнка с момента рождения:
? аутосомные — обусловлены дефектами генов, локализованных в соматических хромосомах, т.е. аутосомах;
? доминантные — развиваются, когда мутация в одном из двух аллелей достаточна для изменения фенотипа, т.е. организм обозначают как гетерозиготу:
? моногенные - обусловлены дефектом одного гена;
? мультифакториальные — имеющие в своей основе как генетическую, так и средовую компоненты;
? рецессивные — развиваются при наличии мутаций в обоих аллелях, т.е. в гомозиготном организме.
• Ген - единица наследственности, определяющая развитие отдельного признака или свойства организма.
• Гетерозигота — особь с двумя структурно различными аллелями (нормальным и мутантным) в определённом локусе.
• Гомозигота - особь с одинаковыми аллелями (нормальными либо мутантными) в определённом локусе.
• Интрон - некодирующая область гена; вырезается в процессе сплайсинга при образовании мРНК из первичного РНК-транскрипта.
• Картирование — локализация элементов генома на генетической карте.
• Клонирование — встраивание чужеродной ДНК в векторную молекулу ДНК или РНК и введение этой конструкции в фаговые, бактериальные или эукариотические клетки хозяина.
• Кодон - последовательность из трёх нуклеотидов в молекуле ДНК или мРНК. соответствующая определённой аминокислоте или сигналу терминации трансляции;
? стоп (нонсенс)-кодон - сигнал терминации (прекращения) трансляции полипептидной цепи (UGA, UAG, UAA);
? инициирующий (стартовый) кодон — AUG-триплет в мРНК, кодирующий метионин, с которого начинается образование полипептидной цепи в процессе трансляции.
• Компаунд - гомозиготная особь, у которой в двух аллелях одного гена есть неодинаковые мутации.
• Комплементарность (нуклеотидных пар) — образование водородных связей по правилу А-Т, Г-Ц в двухнитевой молекуле ДНК.
• Локус — место локализации гена в хромосоме.
• Мутация — изменения в последовательности ДНК:
? делеция — утрата сегмента ДНК размером от одного нуклеотида до субхромосомного фрагмента, включающего несколько генов;
? инсерция вставка сегмента ДНК размером от одного нуклеотида до субхромосомного фрагмента, включающего несколько генов;
? миссенс-мутация (т.е., c изменением смысла) - замена нуклеотида в кодирующей части гена, ведущая к замене аминокислоты в соответствующем белковом продукте;
? нонсенс - замена нуклеотида в кодирующей части гена, сопровождающаяся образованием стоп-кодона;
? сплайсинговая - мутация, затрагивающая сайты сплайсинга или создающая новые сайты сплайсинга в интронных областях гена; сопровождается либо делецией смежного с мутацией экзона, либо невырезанием соответствующего интрона при процессинге первичного РНК-транскрипта;
? точечная - мутация, затрагивающая от одного до нескольких нуклеотидов
• Позиционное клонирование - метод идентификации генов, основанный на молекулярном анализе субхромосомной области их локализации (обратная генетика - от гена к признаку).
• Полимеразная цепная реакция, или специфическая амплификация ДНК, - избирательный синтез in vitro большого числа (порядка миллиона) копий небольшого фрагмента матричной ДНК размером от 50 до нескольких тысяч нуклеотидов; при некоторых условиях возможна амплификация более крупных фрагментов (до 35 тыс. нуклеотидных пар).
• Полиморизм - генетическая изменчивость локуса в определённой популяции.
• Рибонуклоиноная кислота (РНК) - нитевидная молекула, в которой остов из чередующихся остатков рибозы и фосфорной кислоты ковалентно соединен с 4 азотистыми основаниями: аденином, урацилом, гуанином и цитозином.
• Сайт - определённое место в молекуле ДНК.
• Секвенирование - определение нуклеотидной последовательности молекулы ДНК.
• Сплайсинг - процесс вырезания последовательностей, комплементарных интронам, из молекулы первичного РНК-транскрипта.
• Транскрипция — синтез первичных РНК-транскриптов, комплементарных определённым участкам молекулы ДНК (генам).
• Транскрипционный фактор - белок активации или репрессии генной активности, способный взаимодействовать с молекулами ДНК.
• Трансляция - синтез полипептидной цепи по молекуле мРНК.
• Фенотип — совокупность признаков организма, контролируемых определённым генотипом.
• Функциональное клонирование — идентификация генов наследственных болезней начиная с определения первичного биохимического дефекта (прямая генетика — от признака к гену).
• Хромосомы — дискретные внутриядерные структуры, содержащие молекулы ДНК, суперскрученные за счёт взаимодействия с гистоновыми белками.
• Экзон - кодирующий участок гена.
↑ ОСНОВНЫЕ МЕТОДЫ ДИАГНОСТИКИ
Прогресс в доклинической диагностике и понимании молекулярных механизмов сотен монотонных заболеваний и в определении генетических факторов риска развития распространённых болезней человека стал возможен благодаря достижениям молекулярной генетики последних десятилетий. Определённые успехи достигнуты и в изучении механизмов патогенеза мультифакториальных заболеваний, в этиологию которых наряду с факторами внешней среды значительный "вклад" вносят генетические составляющие. Кроме того, достижения геномных технологий позволили выявить монотонно наследуемые формы среди мультифакториальных заболеваний.
Если Вы хотите раз и навсегда избавиться от глазных заболеваний и начать видеть без очков так же как в очках или даже лучше, поправить своё здоровье и научиться управлять Естественными Оздоровительными Функциями Своего Организма, то Вам поможет Майкл Ричардсон, который уже десятилетия преподаёт восстановление здоровья и зрения.
Более тысячи человек уже стали здоровыми, благодаря его методике. Вы готовы взять ответственность за себя и своё здоровье?
Тогда вперёд!
Ваш личный помощник в восстановлении здоровья и зрения, Майкл Ричардсон. Кликните здесь, чтобы начать восстанавливать зрение прямо сейчас!
Более тысячи человек уже стали здоровыми, благодаря его методике. Вы готовы взять ответственность за себя и своё здоровье?
Тогда вперёд!
Ваш личный помощник в восстановлении здоровья и зрения, Майкл Ричардсон. Кликните здесь, чтобы начать восстанавливать зрение прямо сейчас!
До развития геномных технологий генеалогический метод был единственным, позволявшим определить группы риска и его степень для определённого заболевания.
Для мультифакториальных заболеваний, а также в тех случаях, когда затруднено определение типа наследования болезни, его применение дли оценки степени риска имеет меньшее значение, а полученные данные носят ориентировочный, приблизительный характер, и степень их достоверности зависит от полноты генеалогической информации. Методы ДНК-диагностики считают более точными, чем традиционные методы диагностики. Это определяет их незаменимость при проведении медико-генетического консультирования и в развитии предиктивной медицины, так как использование этих методов позволяет точно оценить генетический риск развития заболевания в семье.
Обобщённая и систематизированная информация о локализации и функциях отдельных генов доступна в регулярно обновляемом каталоге наследственных заболеваний человека под названием «Менделевское наследование у человека: каталог человеческих генов и генетических болезней» («Mendelian inheritance in man. Catalogs of autosomal dominant, autosomal recessive and X-linked phenotypes»). Оно содержит сведения о более чем 6500 картированных локусах человека. Для более чем 2800 генов определена их функция.
В настоящее время существует несколько стратегий картирования генов. Первоначально использовали стратегию функционального клонирования, опирающуюся на знание первичного биохимического дефекта или функции гена. С помощью такого подхода были картированы гены, структурные нарушения в которых приводят к развитию распространённых наследственных заболеваний. Однако процесс картирования продвигался медленно, так как патогенез многих наследственных заболеваний и первичный биохимический дефект неизвестны. Первый аутосомный ген человека был картирован в 1968 г. К 1973 г. на хромосомах человека было картировано всего 64 гена, а к 1994 г. было локализовано уже свыше 60 000 маркерных ДНК-последовательностей, в том числе около 5000 структурных генов. Такой прогресс в картировании генов стал возможен благодаря появлению новых технологий и использованию стратегии позиционного клонирования. Эта стратегия основана на информации о локализации гена заболевания на определённой хромосоме или, ещё точнее, в её конкретном сегменте (рис. 23-3).

Однако для большинства генетических заболеваний применяют комбинированный подход, объединяющий функциональное и позиционное клонирование и называемый стратегией позиционного гена-кандидата. На заключительном этапе таких исследований необходимо доказать, что клонированный ген действительно искомый, а нарушение его структуры приводит к заболеванию. Поэтому только обнаружение мутаций в гене-кандидате доказывает его связь с развитием наследственного заболевания.
Различают прямую и косвенную ДНК-диагностику наследственных заболеваний. Прямые методы используют, если ген, нарушение последовательности которого приводит к заболеванию, картирован, известно число его экзонов и интронов, а в идеале и нуклеотидная последовательность. В остальных случаях используют косвенные методы ДНК-диагностики, основанные на использовании сцепленных с геном полиморфных маркёров. Точные молекулярные характеристики каждой мутации могут быть получены только после прямого секвенирования. Однако секвенирование всех экзонов для обнаружения мутаций у каждого отдельного пациента трудоёмко, дорого и требует больших затрат времени. Поэтому на практике сначала более простыми методами проводят предварительный отбор фрагментов ДНК, предположительно содержащих мутации, и в дальнейшем их секвенируют. Методы первичного поиска фрагментов ДНК, предположительно содержащих мутации, основаны на сравнении мутантных и нормальных последовательностей по ряду физических и химических характеристик. Например, небольшие делеции и инсерции приводят к изменению размеров амплифицированных фрагментов ДНК. Эти изменения могут быть зарегистрированы при электрофорезе этих фрагментов в полиакриламидном геле. При точечной мутации гена, т.е. замене одного нуклеотида, длина фрагмента остаётся постоянной, однако изменяются некоторые физико-химические свойства мутантной ДНК.
SSCP-апализ - анализ конформационного полиморфизма однонитевых фрагментов ДНК. Метод основан на регистрации различий в электрофоретической подвижности однонитевых фрагментов ДНК. Различия в скорости миграции разных по нуклеотидному составу однонитевых ДНК при проведении их электрофореза в полиакриламидном геле связаны с образованием этими молекулами различных конформаций, индивидуальность которых зависит от различий и их первичной структуре.
DGGE - денатурирующий градиентный гель-электрофорез основан на зависимости свойств плавления (или денатурации) небольших двухнитевых молекул ДНК от их нуклеотидной последовательности, а точнее от соотношения пар А-Т и G-C в исследуемых фрагментах. При электрофорезе амплифицированных двухнитевых фрагментов ДНК в геле с линейно возрастающей концентрацией денатурирующих веществ плавление нитей ДНК происходит в строго специфичной для данной последовательности области, соответствующей температуре плавления (разьединения нитей ДНК). После начала плавления продвижение двухнитевого фрагмента ДНК в геле резко замедляется.
СМС - метод химического расщепления некомплементарных сайтов основан на способности некоторых химических агентов специфически разрывать нить ДНК в месте локализации неспаренного основания.
НА - метод гетеродуплексного анализа позволяет идентифицировать мутации, находящиеся в компаунде или гетерозиготном состоянии. Метод гетеродуплексного анализа использует преимущество образования гетеродуплексов между двумя разными видами ДНК (мутантной и дикого типа) после их совместного нагревания и медленного охлаждения. При проведении амплификации происходит естественное образование двух типов структур: гомо- и гетеродуплексов. При этом каждый тип структуры представлен двумя видами разных молекул. Присутствие гетеродуплексов в большинстве случаев легко выявляют после проведения электрофореза в полиакриламидном геле.
Наиболее распространённый способ скрининга мутаций — комбинация SSCP - и гетеродуплексного метода, позволяющая выявить точечную мутацию почти в 100% случаев.
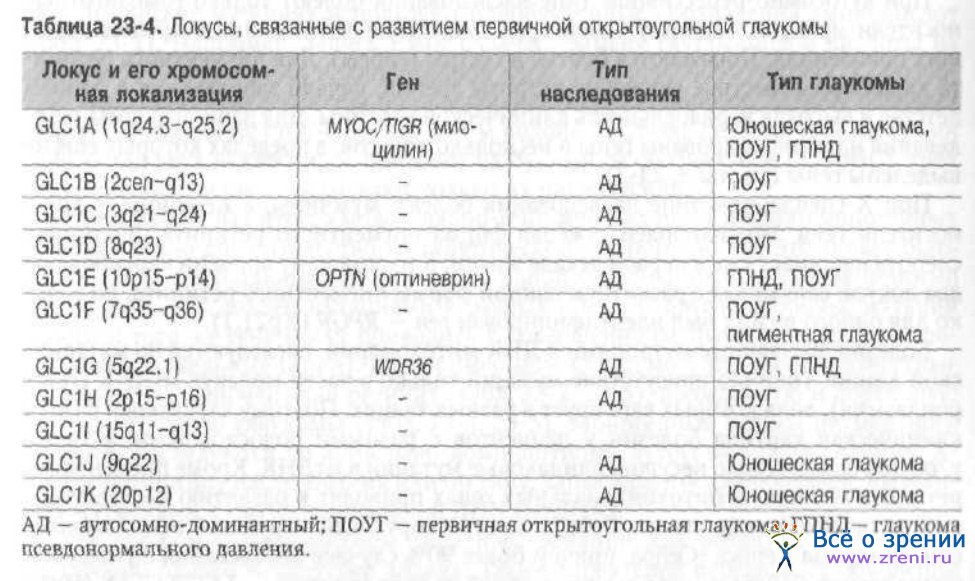
При описании гена указывают его хромосомную локализацию, используя данные о дифференциальной окраске метафазных хромосом. Согласно им каждая хромосома может быть разделена на сегменты, нумерация которых начинается вверх от центромерного района (короткое плечо р) или вниз (длинное плечо q). Для примера возьмём локус GLC1G (5q22.1), ассоциированный с развитием первичной открытоугольной глаукомы. Такая запись означает, что ген (WDR36) локализован в 1-й субъединице 2-го бэнда 2-го сегмента длинного плеча 5-й хромосомы.
↑ НОМЕНКЛАТУРА МУТАЦИЙ
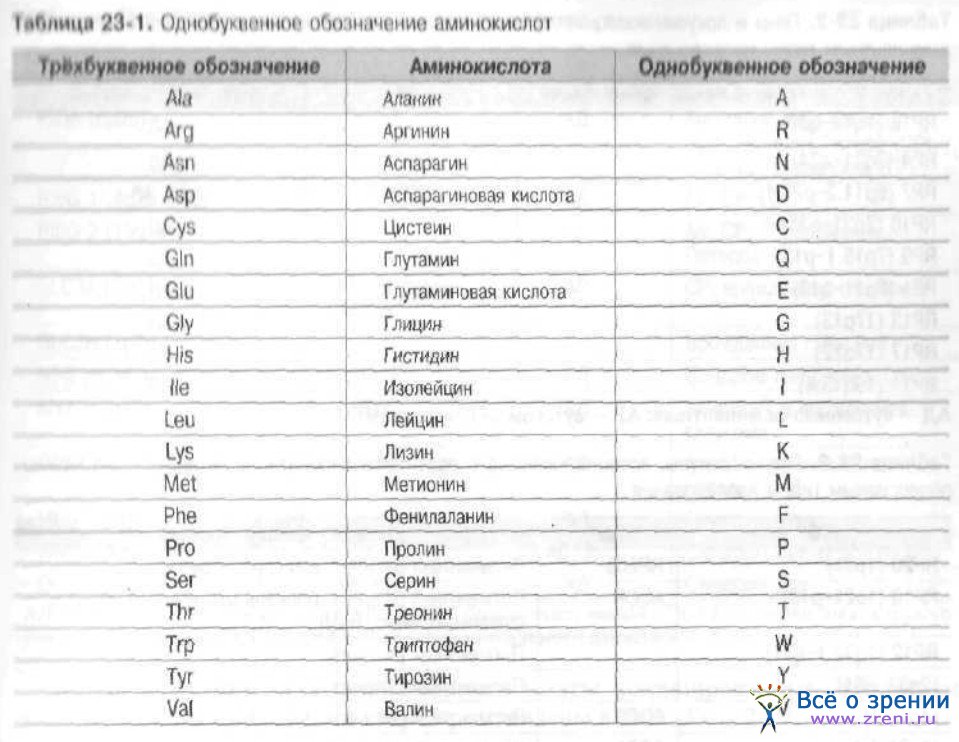
Для описания мутаций и полиморфизмов используют общепринятую номенклатуру, предложенную Артуром Боде и Лап Чи Тсуи в 1992 г. Она рассчитана на запись изменений в нуклеотидной последовательности гена и соответствующих аминокислотных замен в белке. Для обозначения аминокислотных замен при миссенс- и нонсенс-мутациях используют однобуквенные обозначения аминокислот (табл. 23-1).
Например, запись E50K означает замену глутаминовой кислоты на лизин в 50-м положении полипептидной цепи. При нонсенс-мутациях X обозначает место остановки синтеза полипептидной цепи. Например, Q368X означает замену глутамина на стоп-сигнал в 368-м кодоне. В случае делеции или инсерции одного или двух нуклеотидов приводят их буквенное обозначение (555delC), трёх и более нуклеотидов — указывают только их число (850ins7).
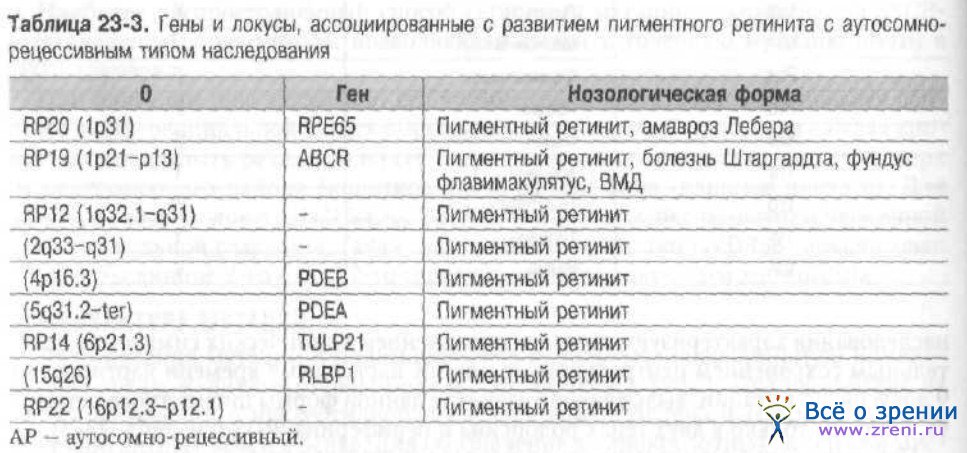
Одним из важных итогов молекулярно-генетических исследований моногенных болезней стало доказательство их генетической гетерогенности. Это может быть обусловлено несколькими причинами. С одной стороны, один и тот же фенотип может быть обусловлен мутациями в разных генах. С другой стороны, мутации одного и того же гена могут приводить к совершенно разным клиническим проявлениям (аллельные серии). Как пример можно рассмотреть хорошо известные заболевания — пигментный ретинит (табл. 23-2,23-3),

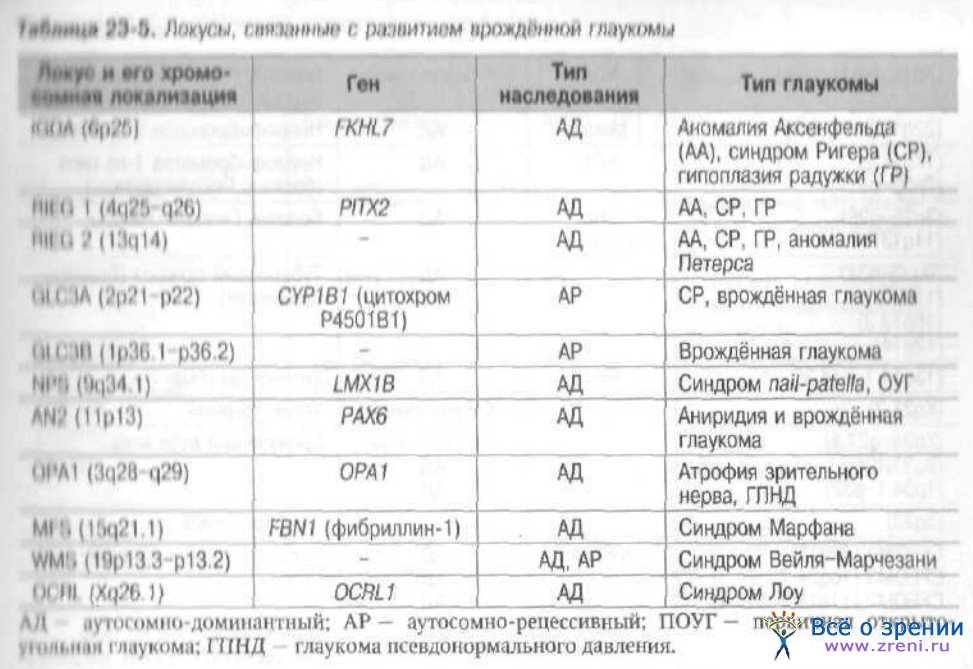
врождённую и первичную открытоугольную глаукомы (табл. 23-4, 23-5).
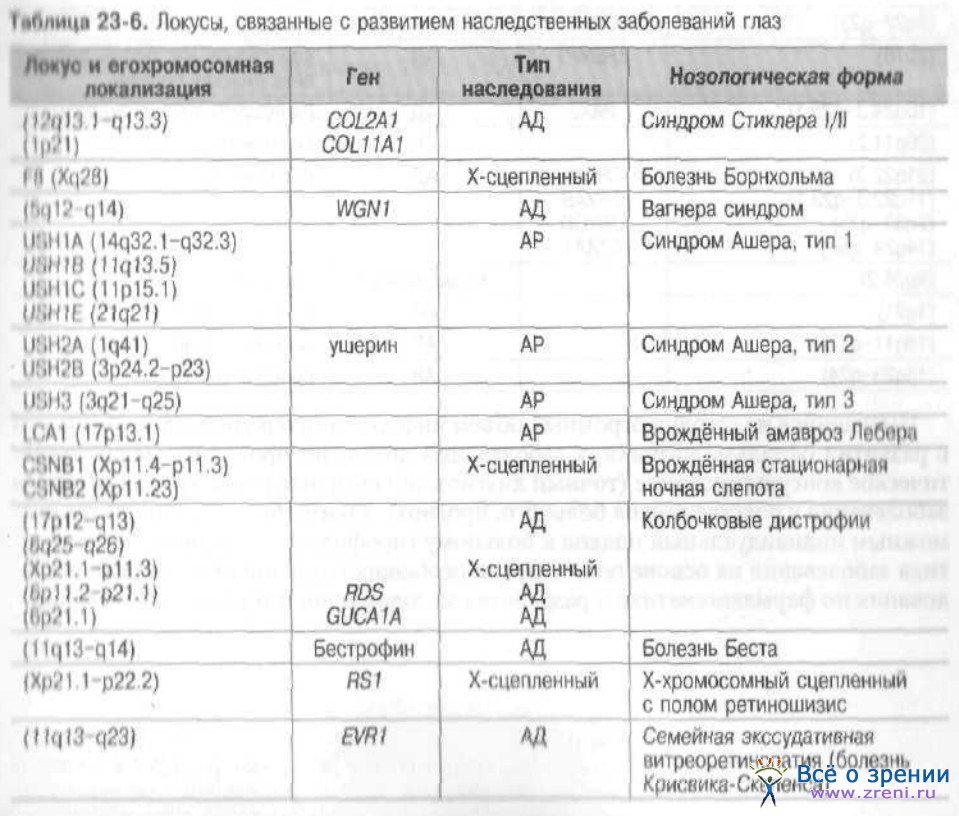
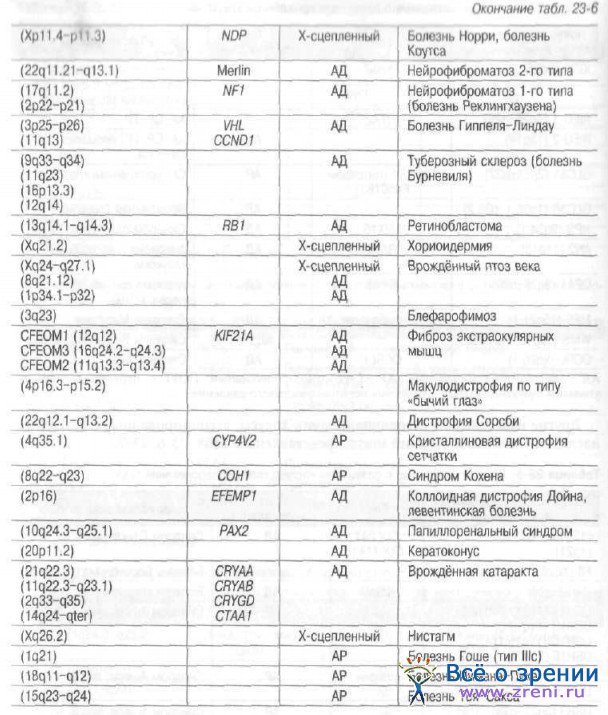
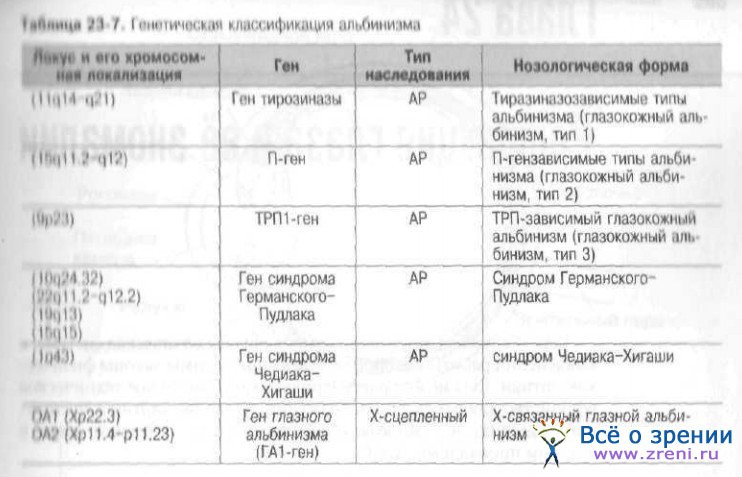
Другие известные на сегодняшний день локусы, ассоциированные с развитием наследственных заболеваний глаза, представлены в табл. 23-6, 23-7.
Моногенные формы пигментного ретинита наследуются по аутосомно-доминантному, аутосомно-рецессивному. X -сцепленному типу наследования или по типу, характерному для болезней, вызванных мутациями в митохондриальной ДНК (мтДНК).
При аутосомно-доминантном типе наследования для развития заболевания достаточно одного мутантного аллеля. Также характерна передача от поколения к поколению, 50% риск развития заболевания в каждом поколении, отсутствие разницы в частоте заболевания между полами. Пигментный ретинит с таким типом наследования характеризуют поздним проявлением клинических симптомов, длительным сохранением центрального зрения. К настоящему времени картировано 9 локусов, но мутации, вызывающие развитие данной формы пигментного ретинита, описаны только в двух генах родопсина и периферина/RDS (см. табл. 23-2).
При аутосомно-рецессивном типе наследования болеют только гомозиготные носители мутации. Поэтому характерно отсутствие заболевания в предшествующих поколениях. Поражаются братья и сёстры (сибсы). Для пигментного ретинита характерны быстрое снижение остроты зрения, начало заболевания в раннем детстве и высокая вариабельность клинической картины. Для данной формы заболевания идентифицированы гены и несколько локусов, в пределах которых ещё не выделены гены (см. табл. 23-3).
При Х-сцепленном типе наследования болеют мужчины, а женщины - лишь носители гена. Это наиболее тяжёлая форма пигментного ретинита. Частичная слепота наступает уже в первой декаде жизни. К настоящему времени картированы два локуса, связанных с развитием данной формы пигментного ретинита, но только для одного из них был идентифицирован ген - RPGR (Хр21.1).
Болезни, вызванные мутациями в ДНК митохондрий, наследуются по материнской линии. Типично присутствие мутаций только в части молекул мтДНК (гетероплазмия). доля которых варьирует в разных тканях. Поэтому характерна разная клиническая картина болезни у пациентов с разными родословными, а также у членов одной семьи, несущих одинаковые мутации в мтДНК. Кроме пигментного ретинита мутации в митохондриальных генах приводят к развитию наследственной оптической нейропатии Лебера, прогрессирующей наружной офтальмоплегии, синдрома Кернса Сейра, причём более 90% случаев заболевания оптической нейропатией Лебера в мире вызвано одной из трёх мутаций - 11778 G ->А (69%) в гене ND4; на две другие мутации в позициях 3460 (ген ND1) и 14484 (ген ND6) приходится 13% и 14% соответственно.
Имеющийся на сегодня огромный объём информации о роли наследственности в развитии офтальмологических заболеваний позволяет проводить медико-генетическое консультирование (точный диагноз, объективная оценка риска развития заболевания у родственников больного, прогноз). Таким образом, становится возможным индивидуальный подход к больному (профилактика, лечение и диагностика заболевания на основе генетических особенностей пациента), а также исследования по фармакогенетике и разработка методов генной терапии заболеваний.
---
Статья из книги: Офтальмология. Национальное руководство | Аветисов С.Э.

Комментариев 0