Болезнь беста


Описание
Болезнь Беста — редко встречающееся двустороннее заболевание, наследуемое по аутосомно-доминантному типу и характеризующееся полиморфными, как правило, асимметричными изменениями в макуле.Заболевание впервые описано в 1883 г. J.E. Adams, который обнаружил «своеобразные» симметричные желтые очаги в макуле обоих глаз у 37-летней женщины. В 1905 г. F. Best сообщил о 8 больных с макулярной дегенерацией из семьи, насчитывающей 59 человек, продемонстрировав, что заболевание передается по аутосомно-доминантному типу. Оно известно в литературе под следующими названиями; «вителлиформная дистрофия Беста», «центральная экссудативная отслойка сетчатки», «наследственные макулярные псевдокисты», «наследственная вителлиформная макулярная дегенерация».
Заболевание встречается с частотой приблизительно 1:10 000 или реже.
Наиболее характерным проявлением болезни Беста являются изменения в макуле диаметром от 0,3 до 3 РД, вызывающие ассоциации с яичным желтком.
Генетические исследования. Болезнь Беста — заболевание с аутосомно-доминантным типом наследования и высокой пенетрантностью. В 1992 г. было установлено, что ген, вызывающий развитие болезни Беста, локализуется в 11-Й хромосоме. В последующих исследованиях ген, ответственный за развитие вителлиформной макулодистрофии, был картирован на длинном плече 11-й хромосомы в интервале между маркерными генами D11S903 и PYGM (ген, кодирующий мышечную гликогенфосфорилазу) или, как уточнили позднее, между маркерными генами D11S986 и DI1S480. Этот ген кодирует синтез белка, названного бестрофин. Кроме того, в данной области был картирован ген ROMІ, кодирующий фоторецептор-специфический мембранный белок. В связи с этим было высказано предположение, что болезнь Беста является результатом мутаций, возникающих в гене ROMI, картировав ген ROMІ, а также локусы D11S986, UGB (ген утероглобина) и PYGM, показали, что UGBлокализуется проксимально по отношению к ROMІ и оба гена рекомбинируют при болезни Беста. Авторы сделали вывод, что ген R0M1 не вызывает развития болезни Беста.
В 1998 г. К. Petrukhin и соапт. идентифицировали ген VMD2, являющийся причиной болезни Беста в 5 семьях, в области 1 lqI3. Авторы обнаружили пять различных мутаций ядерной ДНК в экзонах 2, 4, 6 и 8-го гена VMB2.
Область llql3—14, в которой локализуется ген VMD2, перекрывающая приблизительно 40 сантим орган ид, печально известна офтальмогенетикам как «прибежище» множества генов, ответственных за развитие различных заболеваний сетчатки, в частности глазокожной формы альбинизма, синдрома Ушера, синдрома Барде—Билля, аутосомно-доминантной формы семейной экссудативной витреоретинопатии.
R.E. Ferrell и соавт. (1983) при обследовании пациентов с атипичной семейной вителлиформной макулодистрофией выявили ее связь с классическим маркером GPT-1, локализующимся на длинном плече 8-й хромосомы. У некоторых больных из обследованной ими семьи была нормальная ЭОГ, а диаметр очагов вителлиформных изменений в макуле во всех случаях не превышал I РД.
Фенотипическая вариабельность болезни Беста может объясняться генетической гетерогенностью заболевания и, помимо этого, может быть обусловлена неизвестными ДО настоящего времени мутациями. S. Nordstrom и W. Thorbum (1980) сообщили о семье, в которой у гомозиготного отца было 11 дочерей с различными клиническими проявлениями болезни Беста. Известно около 10 различных мутаций гена VMD2, вызывающих развитие болезни Беста у детей и фенотипически похожих на нее макулопатий у взрослых.
Гистологические и гистохимические исследования. В ходе исследований с использованием световой и электронной микроскопии установлено, что в ранних стадиях болезни Беста клетки пигментного эпителия поглощаются цитосомами с аномальной не идентифицируемой субстанцией. Между клетками пигментного эпителия сетчатки и нейроэпителия накапливаются гранулы липофусциноподобного вещества, нарушаются целостность мембраны Бруха и структура нервных элементов сетчатки: большая часть наружных сегментов фоторсцепторов исчезает или дегенерирует, при этом во внутренних сегментах фоторецепторов пораженных участков сетчатки происходит накопление кислых мукополиса- харвдов. Капилляры проникают в субретинальное пространство через измененную мембрану Бруха.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Во П стадии внутри клеток пигментного эпителия сетчатки в макуле происходит накопление значительного количества гранул вещества, гистохимически близкого липофусцину, а также макрофагов в субретинальном пространстве и в хороидее. В поздней фазе заболевай и к отмечается упрощение клеток и в макуле. Диаметр клеток превышает норму в 1,5—2 раза.
У больных с вителлиформной дистрофией старшего и пожилого возраста диффузные отложения липофусциноподобного вещества были выявлены в слое пигментного эпителия, во внутренних сегментах фоторецепторов, в клетках Мюллера и даже в стекловидном теле.
Классификация. В зависимости от офтальмоскопических проявлений выделяют 5 стадий в течении болезни Беега:
стадия 0 — изменений в макуле нет, но регистрируется аномальная ЭОГ ;
стадия I — минимальные пигментные нарушения и очажки гипопигментации в макуле, видимые при ФАГ как окончатые дефекты;
стадия II — классическая вителлиформная киста в макуле;
стадия III — разрыв кисты и различные фазы резорбции ее содержимого (псевдогипопион);
стадия IV — оранжево-красные атрофические изменения в макуле после резорбции содержимого кисты (IVa), формирование фиброглиального рубца (IV6), субретинальная неоваскуляризация (lVc).
Классификация по стадиям заболевания недостаточно корректна, так как нет убедительных доказательств последовательной эволюции макулярных изменений через описанные выше стадии у большинства пациентов с болезнью Беста .
Клинические проявления. У многих пораженных членов семей с болезнью Беста глазное дно выглядит нормальным. Иногда отмечают ослабление фовеального рефлекса. Даже в возрасте 40—50 лет у 5 % пациентов с болезнью Беста при офтальмоскопическом обследовании не обнаруживается изменений на глазном дне. Диагностировать заболевание в подобной ситуации можно только при помощи электроокулографии (ЭОГ). У пациентов с болезнью Беста при ЭОГ регистрируют снижение коэффициента Ардена — отношения светового пика к темновому спаду.
Течение болезни Беста обычно бессимптомное. При наличии офтальмоскопических проявлений заболевание выявляют случайно при осмотре детей в возрасте 2—6 лет. Макулярные изменения в большинстве случаев двусторонние и, как правило, асимметричные. Для 1 (превителлиформной) стадии болезни Беста характерно появление мелких желтых пятнышек в макуле. У некоторых пациентов в макуле определяют глыбки пигмента, разделенные мелкими желтыми очажками липофусциноподобного вещества и участками депигментации, которые напоминают изменения при ретикулярной дистрофии.
Во II стадии болезни размеры вителлиформных кист, напоминающих «яичный желток», могут достигать диаметра от 0,3 до 3 РД (рис. 5.1).
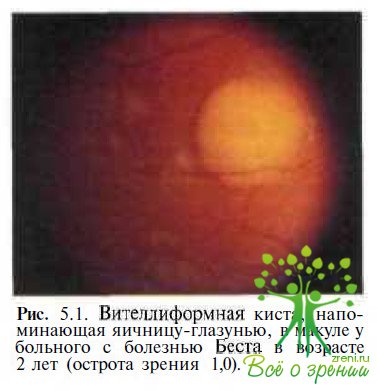
Несмотря на выраженные изменения в макуле острота зрения у пациентов в этой стадии болезни обычно составляет 0,5—0,9. Манифестация болезни в виде вителлиформных кист может варьировать в широком возрастном диапазоне — типичная картина «яичницы-глазуньи» описана у 7-дневного ребенка и у мужчины в возрасте 62 лет.
Снижение остроты зрения обычно происходит в III стадии заболевания, когда отдельные вителлиформные кисты разрываются, вызывая ассоциацию с «яичницей-болтушкой». Позднее в результате частичной резорбции и смещения липофусциноподобного содержимого в нижнюю половину кисты последняя напоминает картину «пеевдогипопиона» (рис. 5.2).
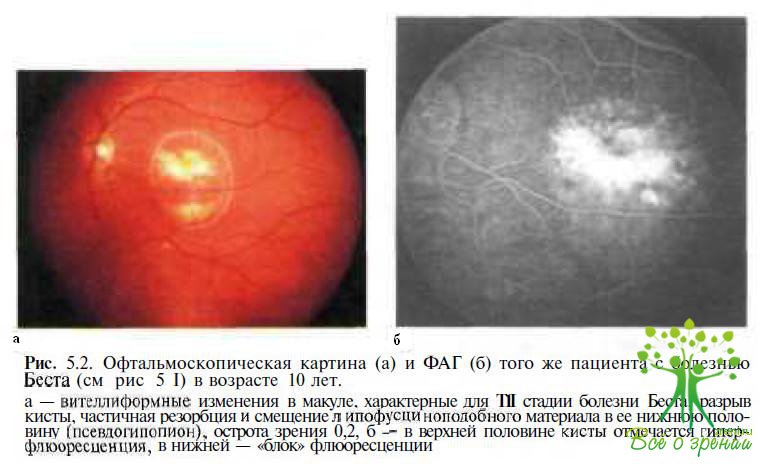
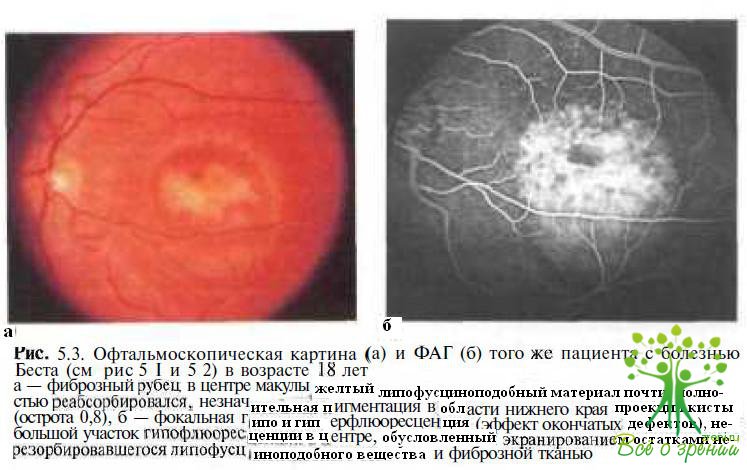
В дальнейшем по мере резорбции липофусциноподобного вещества формируется фиброзный рубец (рис. 5.3) или, несколько реже, субретинальная неоваскулярная мембрана.
Нередко у детей с болезнью Беста субретинальную неоваскуляризацию определяют в раннем возрасте. Мы наблюдали развитие субретинальной неоваскулярной мембраны левого глаза у 6-летней девочки (рис 5.4).

Интересно, что при предшествующем наблюдении у этой девочки не было отмечено развития вителлиформной кисты в макуле левого глаза, а отмечались только двусторонние желто-оранжевые атрофические изменения и точечные скопления липофусциноподобного вещества по краям очага (см. рис. 5.4, а). Субретинальную неоваскуляризацию отмечают приблизительно у 5 % пациентов с болезнью Беста.
В любой стадии заболевания у пациентов с болезнью Беста могут возникать субретинальные кровоизлияния. S.A. Miller и соавт. (1976) сообщили о 9-летнем мальчике, у которого были обнаружены интактная вителлиформная киста справа и вскрывшаяся киста с частично резорбировавшимся содержимым, субретинальными геморрагиями и неоваскулярной мембраной слева.
А.Р. Schachat и соавт. (1985) наблюдали казуистический случай — формирование макулярного разрыва и регматогеиной отслойки сетчатки у патента с болезнью Беста. В преклонном возрасте у больных вследствие утраты хориокапилляров и атрофии пигментного эпителия сетчатки нередко развивается хориоидальный склероз в макулярной области.
В литературе имеются сообщения о множественных вителлиформных поражениях при болезни Беста. В этих случаях макулярные вителлиформные изменения сочетаются с экстрафовеальными поражениями сетчатки, локализующимися преимущественно вдоль верхневисочной сосудистой аркады. Обычно насчитывается от 2 до 5 экстрамакулярных вителлиформных поражений. Их диаметр несколько меньше, чем размеры макулярных изменений, и не превышает 0,2-1 РД. Поражения-сателлиты могут эволюционировать асинхронно по отношению друг к другу и изменениям в макуле, а могут находиться на той же стадии развития.
Флюоресцентная ангиография. Антиографическая картина при болезни Беста варьирует в зависимости от стадии заболевания. В превителлиформной стадии заболевания в зонах атрофии пигментного эпителия в макуле выявляют мелкие окончатые дефекты, обусловливающие локальную гиперфлюоресценцию. В вителлиформной стадии отсутствует флюоресценция в границах кисты (так называемый блок флюоресценции), после разрыва кисты (стадия псевдогипопиона) — гиперфлюоресценция в ее верхней половине, а в нижней сохраняется «блок» флюоресценции (см. рис. 5.2, б). После частичной резорбции липофусциноподобного содержимого кисты в макуле выявляют окончатые дефекты (см. рис. 5.3, б).
Зрительные функции. Жалобы. Пациенты с болезнью Беста обычно предъявляют жалобы на затуманивание зрения, снижение остроты зрения, затруднения при чтении текстов с мелким шрифтом и метаморфопсии. Метамopфопсии, вероятно, обусловлены неровностями макулярного рельефа из-за субретинальных скоплений липофусциноподобного вещества, пигмента и рубцовых изменений.
Острота зрения. Острота зрения у детей с болезнью Беста варьирует в зависимости от стадии заболевания от 0,02 до 1,0. Изменения на глазном дне не соответствуют функциональным нарушениям. Даже у детей с вителлиформной кистой или после ее разрыва острота зрения в течение многих лет может составлять 0,8—1,0. Нередко отмечают флюктуации остроты зрения в пределах 1—2 строк таблицы, не сопровождающиеся изменениями на глазном дне.
Мы наблюдали в течение 18 лет мальчика с болезнью Беста, у которого в возрасте 1,5 лет была выявлена вителлиформная киста (см. рис. 5.1). В течение 8 лет острота зрения ребенка варьировала от 0,9 до 1,0. В возрасте 10 лет после разрыва кисты и смещения липофусциноподобного содержимого в ее нижнюю половину с формированием псевдогипопиона острота зрения у ребенка снизилась до 0,2 (см. рис. 5.2), но впоследствии восстановилась до 1,0 и не изменялась до последнего обследования, проведенного в возрасте 19 лет (см. рис. 5.3).
При длительном наблюдении за 58 пациентами с болезнью Беста в различных стадиях R. Clement (1991) установил, что у 43 % из них острота зрения составляла 0,5 и выше. G.A. Fishman и соавт. (1993), обследовав 47 пациентов с болезнью Беста во II—IV стадиях в возрасте 5—72 лет из 23 семей, обнаружили, что у 41 % из них острота зрения была не ниже 0,3. Между тем лишь у 20 % пациентов старше 40 лет зрение соответствовало требованиям водительской комиссии, а у всех пациентов старше 50 лет с офтальмоскопическими изменениями, соответствующими III—IV стадиям болезни, острота зрения лучше видящего глаза не превышала 0,1—0,3.
D.W. Park и соавт. (1999) при обследовании 107 больных установили, что у лиц моложе 30 лет средняя острота зрения составляла U,H„ в возрасте 30-60 лет - 0,65, старше 60 лет - 0,5.
Цветовое зрение у большинства больных нормальное. Темповая адаптация не изменяется. Пространственная контрастная чувствительность у детей с остротой зрения 0,5 и выше обычно снижена в области высоких пространственных частот.
Периферические границы поля зрения сохраняются нормальными. Порог яркостной чувствительности в превителлиформных стадиях болезни у пациентов не изменяется. При компьютерной статической периметрии у пациентов с разорвавшейся вигеллиформной кистой и сниженной остротой зрения может быть выявлена относительная центральная скотома.
Электрофизиологические исследования. Единственным достоверным тестом, позволяющим диагностировать болезнь Беста даже у детей с отсутствием офтальмоскопических изменений, является ЭОГ. Проведение ЭОГ возможно у детей в возрасте 6 лет и старше. Характерными признаками болезни Беста являются низкий базовый потенциал и снижение коэффициента Ардена — отношения светового пика к темновому спаду. Коэффициент Ардена у пациентов с болезнью Беста не превышает 1,5 (150 %). Общая и локальная ЭРГ при болезни Беста долго сохраняются нормальными.
Дифференциальная диагностика. Диагноз у пациентов с болезнью Беста устанавливают на основании офтальмоскопической картины и результатов регистрации ЭОГ и ЭРГ. В трудных случаях помощь в диагностике могут оказать молекулярные генетические исследования, а также офтальмоскопическое и электрофизиологическое обследования других членов семьи.
Болезнь Беста необходимо дифференцировать от отслойки пигментного эпителия, болезни Коатса, острого хориоретинита различной этиологии, других макулярных дистрофий пигментного эпителия сетчатки (бабочковвдной дистрофии, Х-образной макулярной дистрофии), болезни Штаргардта, вителлиформной дистрофии взрослых.
Отслойка пигментного эпителия. Сложности в диагностике возникают у пациентов с атипичной клинической картиной и экссудативными изменениями в макуле (рис. 5.5).
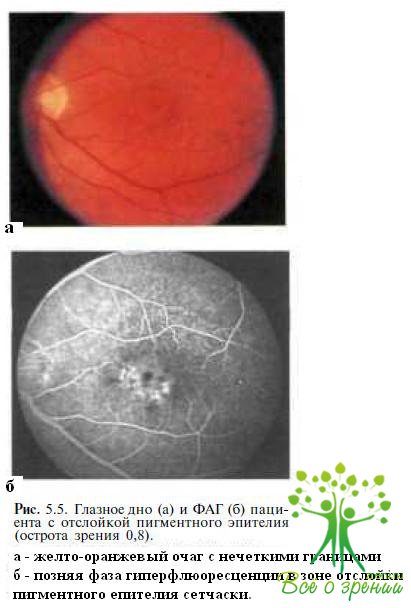
Заболевание чаще одностороннее. Общая ЭРГ не изменяется, локальная ЭРГ может быть снижена. ЭОГ в большинстве случаев нормальная. При ФАГ в поздней фазе у пациентов с отслойкой пигментного эпителия сетчатки выявляют гиперфлюоресценцию (см. рис. 5.5, б).
Болезнь Коатса. У большинства больных с ретинитом Коатса во время первого обследования у офтальмолога в макуле выявляют проминирующий очаг твердого ексудата желтого песта (рис. 5.6),
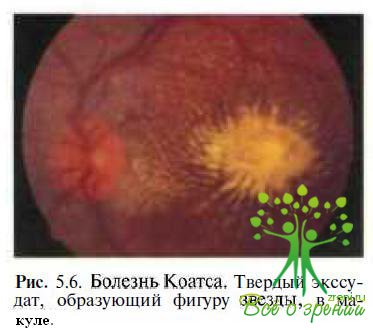
напоминающий вителлиформные изменения при болезни Беста. Экссудативные изменения в макуле нередко сочетаются с субретинальными геморрагиями и неоваскуляризацией. Диагноз основывается на результатах офтальмоскопии: при осмотре периферии глазного дна с использованием налобного офтальмоскопа и линз 20—30 дптр или биомикроекопии с трехзеркальной линзой Гольдмана у всех пациентов с болезнью Коатса выявляют сосудистые аномалии (телеангиэктазии, микро- и макроаневризмы, артериовенозные шунты), чаще локализующиеся в височной половине сетчатки. Заболевание спорадическое. У 95 % больных поражается один глаз. ЭОГ в начальной и развитой (Па) стадиях болезни не изменяется. Острота зрения у пациентов с ретинитом Коатса при наличии экссудативных изменений в макуле не превышает 0,4.
Острый токсоплазмозный хориоретинит. Диагностические трудности возникают обычно при обследовании пациентов с приобретенной формой токсоплазмоза и экссудативно-геморрагическими изменениями в макуле, имитирующими вителлиформные очаги с субретинальными кровоизлияниями (рис. 5.7).
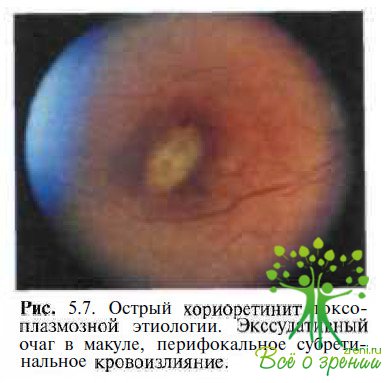
Заболевание обычно одностороннее. Хориоретинальные поражения сочетаются с витреитом различной тяжести, а иногда и изменениями переднего отрезка. Пациенты обычно жалуются на внезапное значительное снижение остроты зрения, которая в большинстве случаев варьирует от 0,01 до 0,2. При статической периметрии выявляют повышение порога яркостной чувствительности, абсолютную или относительную центральную скотому. ЭОГ не изменена. Подтвердить диагноз токсоплазмоза позволяют положительные результаты иммунологических исследований.
Болезнь Штаргардта. Заболевание передается по аутосомно-рецессивному типу. Острота зрения снижается до 0,1 и ниже уже в возрасте 6—10 лет. При офтальмоскопии определяют различной выраженности атрофические изменения в макуле и/или множественные мелкие пятна бело-желтого цвета в полюсе и на периферии. При ФАГ в макуле регистрируют перифовеальные участки гиперфлюоресценции на фоне гипофлюоресценции окружающей хороидеи.
В трудных случаях для дифференциальной диагностики используют электрофизиологические исследования: у большинства детей с болезнью Штаргардта амплитуда скотопической и фотопической ЭРГ сохраняется нормальной, локальная и ритмическая ЭРГ снижены, а параметры ЭОГ изменены на 20—80 % по сравнению с нормой.
Mакулярные дистрофии пигментного эпителия сетчатки. Дифференциальная диагностика болезни Беста и макулярных дистрофий пигментного эпитерндистрофий) вызывает наибольшие трудности. В литературе описаны большие семьи, у некоторых членов которых выявляли макулярные лиформные изменения, а у других — нарушения, напоминающие паттерндистрофии (бабочковидную, ретикулярную или Х-образную макулярные дистрофии). Известны случаи сочетания вителлиформных и паттернподобных изменений в макуле правого и левого глаз у одного пациента а также их комбинация с одной стороны. D.W. Park и соавт. (1999) наблюдали такие сочетанные изменения у пациентов с болезнью Беста, верифицированной при помощи молекулярных генетических исследований. В то же время вителлиформизменения были обнаружены у больного с мутацией гена RDS/peripherin (Gly- 167-Asp). Известно, что мутации гена RDS/peripherin были установлены у пациентов с паттерн-днетрофиями , аугосомно-доминантной формой пигментного ретинита. Таким образом, офтальмоскопически не всегда можно различить болезнь Беста и паттерндистрофии. Для дифференциальной диагностики в таких случаях необходимо использовать ЭОГ и ЭРГ. ЭОГ у пациентов с паттерндистрофиями обычно бывает нормальной, реже — измененной. Амплитуда ЭРГ — нормальная или субнормальная.
Лечение. Патогенетически обоснованного лечения нет. При случайном выявлении у пациента болезни Беста необходимо офтальмологическое обследование всех членов его семьи. Представляется целесообразным информирование пациентов о вариабельности течения заболевания и функционального прогноза.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0