Семейная экссудативная витреоретинопатия


Описание
Семейная экссудативная витреоретинопатия (болезнь Крисвика—Скепенса) — генетически гетерогенное двустороннее заболевание, характеризующееся нарушениями васкуляризации сетчатки, фиброзными изменениями в стекловидном теле и частым развитием отслойки сетчатки.Заболевание впервые описано в 1969 г. V. Criswick и С. Schepens,, которые наблюдали своеобразные витреоретинальные изменения у 6 детей из двух семей. Все дети родились в срок с нормальной массой тела, их анамнез не содержал сведений о перинатальной асфиксии или оксигенотерапии в неонатальном периоде. Основными симптомами заболевания были задняя отслойка стекловидного тела, витреальиые преретинальные мембраны, располагавшиеся в заднем полюсе и на периферии, наличие твердого интра- и субретинального экссудата, а также витреоретинальные тракции во всех квадрантах, вызывавшие гетеротопию макулы.
В 1971 г. J. Gow и G.L. Oliver сообщили о 22 больных из трех поколений одной семьи. Проанализировав семейный анамнез, авторы обнаружили сведения о нескольких членах этой родословной со схожей патологией глаз еще в трех предшествовавших поколениях и предположили, что заболевание наследуется по аутосомно-доминантному типу.
Генетические исследования. Патогенез заболевания изучен недостаточно. Предполагают, что генетический дефект, лежащий в его основе, препятствует нормальной индукции васкулогенеза или нарушает дифферениировку фоторецепторов, влияя таким образом на последующие процессы формирования сосудов.
Заболевание обычно передается по аутосомно-доминантному типу. Известно, что ген EVR1, ответственный за передачу аутосомно-доминантной формы семейной экссудативной витреоретинопатии, локализуется на длинном плече 11-й хромосомы в интервале ql3—23, непосредственно примыкая к маркерному гену D1JS873.
Предположение о том, что генетический дефект в области длинного плеча 11-й хромосомы приводит к нарушениям васкуляризации периферических отделов сетчатки, получило еще одно подтверждение в исследовании Н. Uto и соавт. (1994), сообщивших о младенце с I lq-синдромом, у которого были выявлены двусторонние изменения ретинальных сосудов, клинически напоминающие симптоматику при семейной экссудативной витреоретинопатии.
М.А. Samashmus и соавт. (2000) обследовали 16 пациентов с аутосомно-доминантной семейной экссудативной витреоретинопатией из 4 поколений большой семьи. Несмотря на характерные фенотипические проявления, данная форма заболевания генетически отличалась от ранее известных и не была связана с геном EVR1 в локусе llq13.
B.S. Shastry и соавт. (1994) описали родословную с аутосомно-рецессивным типом наследования семейной экссудативной витреоретинопатии, у членов которой при анализе ДНК не было обнаружено изменений в локусах 1 iql3 иХр1 1.3. G. de Crecchio и соавт. (1998) сообщили об аутосомно-рецессивном тине наследования экссудативной витреоретинопатии у пациентов из двух итальянских семей.
Имеются сведения о существовании родословных с Х-сцепленным типом наследования семейной экссудативной витреоретинопатии. Установлено, что у больных семейной экссудативной витреоретинопатией с Х-сцепленным типом наследования генетический дефект локализуется на Х-хромосоме в областях Xq21.3 или Хр11.4 — р11.З. У пациентов с семейной экссудативной витреоретинопатией из родословных с Х-сцепленным типом наследования были выявлены мутации в гене, ответственном за развитие болезни Норри. На основании этих фактов было высказано предположение, что семейная экссудативная витреоретинопатия и болезнь Норри являются аллельными заболеваниями. Описаны также спорадические случаи экссудативной витреоретинопатии.
Ген, ответственный за передачу некоторых витреоретинальных заболеваний, в частности семейной и спорадической экссудативной витреоретинопатии, а также болезни Норри, картирован на проксимальном коротком плече Х-хромосомы. Он кодирует сравнительно небольшой белок, состоящий из 133 аминокислот. Детально функции этого белки пока не изучены, хотя изпсстни гомологичные полипептиды, принимающие участие в процессах клеточной интеграции и дифференцировки. С целью исследования патологических процессов, связанных с этим геном, была создана экспериментальная модель на мышах. Обнаружено, что ген экспрессируется у мышей в слое нервных волокон сетчатки, мозжечке и ольфакторном эпителии. У мутантных мышей появляются помутнения в стекловидном теле, нарушается формирование ганглиозных клеток сетчатки, в некоторых случаях развивается дегенерация фоторецепторов. При регистрации ЭРГ у них отмечается значительное снижение амплитуды Ъ-волны.
Х-сцепленныс формы семейной экссудативной витреоретинопатии характеризуются более тяжелым течением, чем варианты с аутосомно-доминантной передачей. В свою очередь G. de Grecchio и соавт. (1998) отметили, что семейная экссудативная витреоретинопатия, наследуемая по аутосомно-рецессивному типу отличается от аутосомно-доминантной формы заболевания ранней манифестацией и более грубыми фенотипическими проявлениями.
Гистологические исследования. При исследовании глаз с тотальной отслойкой сетчатки обнаружено, что ее внутренняя поверхность в височной половине кзади от зубчатой линии покрыта фиброваскулярной соединительной тканью, закручивающей сетчатку в плотные складки. L.C. Glazer и соавт. (1995) исследовали эпиретинальные мембраны, удаленные у двух пациентов с семейной экссудативной витреоретинопатией, используя световую и электронную микроскопию. Обе мембраны содержали множественные астроциты, располагавшиеся вдоль миофиброиигов. Признаков фенестрации эндотелия сосудов авторы не обнаружили, что не согласуется с офтальмоскопическими и ангиографическими проявлениями заболевания.
В других исследованиях отмечают адгезию сетчатки и проминирующих преретинальных или витреальных мембран с образованием ретинальных складок, наличие интра- и субретинального экссудата, некроз и острое воспаление в периферических отделах сетчатки.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Клинические проявления. При семейной экссудативной витреоретинопатии всегда поражаются оба глаза, но изменения, как правило, асимметричны и могут развиваться со значительной разницей во времени. В легких случаях заболевание протекает бессимптомно, и только тщательное офтальмоскопическое исследование с применением флюоресцентной ангиографии позволяет обнаружить аваскулярные зоны на крайней периферии сетчатки. В тяжелых случаях наиболее постоянные симптомы семейной экссудативной витреоретинопатии — задняя отслойка стекловидного тела, витреоретинальные фиброваскулярные и экссудативные изменения. У пациентов с легкими формами заболевания изменения в стекловидном теле отсутствуют. СЕ. van Nouhuys (У991) при обследовании 90 больных из 16 семей не обнаружил витреальных нарушений у 44 % из них.
J. GOW и G.L. Oliver (1971) выделяют 3 стадии в течении семейной экссудативной витреоретинопатии.
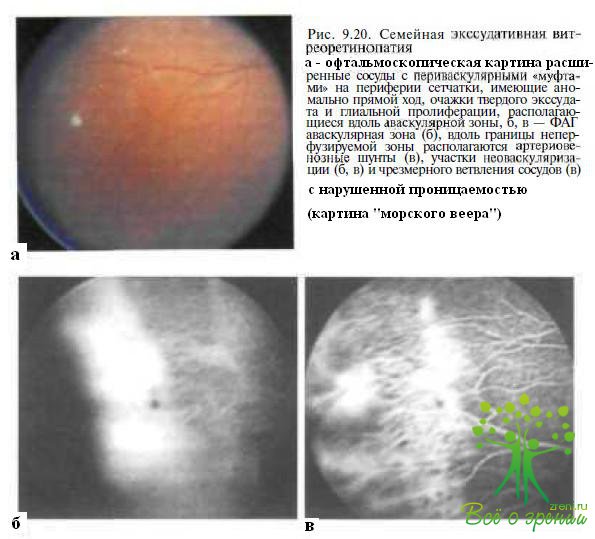
Стадия I характеризуется асимптоматическим течением — при обследовании у больных выявляют периферические аваскулярные зоны, более выраженные в височной половине сетчатки и в некоторых случаях захватывающие всю (до 360°) периферическую сетчатку. При семейной экссудативной витреоретинопатии периферические аваскулярные зоны в большинстве случаев длительное время не претерпевают каких-либо изменений в отличие от ретинопатии недоношенных, при которой, как правило, отмечают регрессию заболевания, проявляющуюся постепенной васкуляризацией неперфузируемых ранее областей сетчатки. Вдоль границы аваскулярной зоны выявляют непролифератннныс изменения: сосуды, идущие от диска, по мере их ветвления приобретают необычно прямой ход и следуют почти параллельно, увеличивается ветвление сосудов на границе с аваскулярной зоной, определяются расширенные и склерозированные сосуды, периваскулярные муфты, точечные очажки твердого экссудата, локализующиеся центральное неперфузируемых зон (рис. 9.20). Происходят незначительные изменения в макуле — сглаженность или исчезновение фовеолярного и макулярного световых рефлексов.
W.E. Benson (1995) при обследовании 39 больных из 22 семей обнаружил изменения, характерные для I стадии, в 44 % (34 из 78) глаз. S.D. Pendergast и М.Т. Trese (1998) выявили аваскулярные зоны без признаков экстраретинальной пролиферации лишь в 12 % (6 из 52) глаз.
При обычной офтальмоскопии обнаружить аваскулярные зоны достаточно сложно. Для их выявления лучше применять непрямую офтальмоскопию с использованием налобного офтальмоскопа с зеленым светофильтром.
Стадия ІІ характеризуется пролиферативными и экссудативными изменениями. При офтальмоскопии выявляют кистовидный макулярный отек, дилатацию и извитость периферических сосудов, интра- и субретинальную экссудацию вдоль границы очагов пролиферации (рис 9.21),

неоваскуляризацию сетчатки на границе аваскулярной и перфузируемой областей, конусообразную экстраретинальную фиброваскулярную пролиферацию и отслойку сетчатки (рис. 9.22).
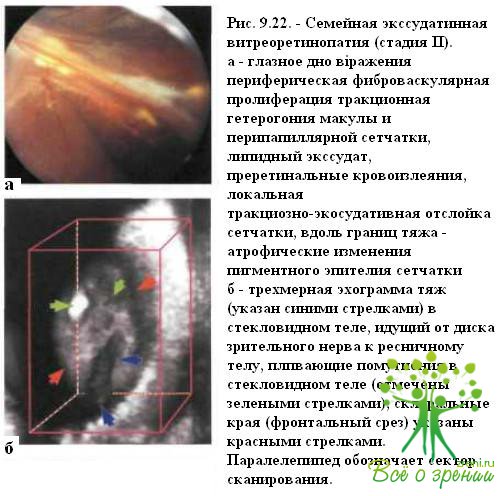
Вокруг зон неоваскуляризации и пролиферации нередко определяются участки гиперпигментации, связанные с вторичными изменениями пигментного эпителия сетчатки (см. рис. 9.22).
Интра- и субретинальная экссудация развивается у 9—22 % бальных, конусообразная экстраретинальная фиброваскулярная пролиферация — у 17—20 %. Нерегматогенная отслойка сетчатки, обычно локализующаяся между экватором и зубчатой линией, происходит во II стадии болезни у 15—17 % пациентов.
Периферическая неоваскуляризация была обнаружена СЕ. van Nouhuys (1991) в 18 из 170 обследованных глаз, причем у половины пациентов она сочеталась с зонами фиброваскулярной пролиферации, располагавшимися центральнее неперфузируемых областей в нижненаружном квадранте и обусловленными ими тракционными деформацией диска зрительного нерва и височной гетеротопией макулы (рис. 9.23).
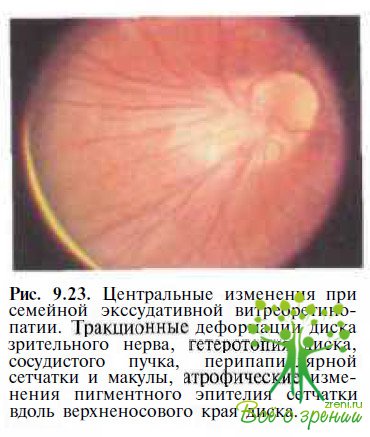
Деформированный диск зрительного нерва обычно слегка проминирует. Тракпионная гетеротопия сетчатки в височную сторону различной степени выраженности наблюдается в 18—49 % глаз. У 8—39 % больных формируется серповидная складка сетчатки, идущая от диска зрительного нерва через задний полюс к периферии в нижненаружном квадранте. У некоторых пациентов с деформированной и смешенной макулой отмечаются миопия и псевдоэкзотропия.
Стадия III характеризуется развитием широкого спектра осложнений, таких как тракционная или регматогенная тотальная отслойка сетчатки, массивная интра- и субретинальная экссудация, лентовидная дистрофия роговицы, вторичная глаукома, осложненная катаракта. Иногда у пациентов с семейной экссудативной витреоретинопатией и отслойкой сетчатки развивается гемофтальм .
Тотальную отслойку сетчатки, обусловленную витреоретинальными тракциями и/или разрывами сетчатки, выявляют у 12—26 % пациентов. Разрывы обычно формируются около неперфузируемых зон в височной половине сетчатки. Тракиионная отслойка сетчатки развивается у 6—10 % больных, в основном в возрасте до 10 лет. Регматогенеза отслойка сетчатки возникает чаще — приблизительно у 25 % больных, как правило, в возрасте от 20 до 30 лет. Экссудатйвная отслойка сетчатки у пациентов с семейной экссудативной витреоретинопагией развивается довольно редко.
Зрительные функции. Острота зрения варьирует в широких пределах — от полной слепоты до 1,0. У пациентов с I стадией заболевания острота зрения обычно нормальная. Снижение остроты зрения во II стадии заболевания происходит вследствие тракционной гетеротопии перипапиллярной сетчатки и макулы в височную сторону. При визометрии у 26 детей с семейной экссудативной витреоретинопатией S.D. Pendergast и М.Т. Trese (1998) отметили остроту зрения 0,03—0,5 в 12,5 % глаз, реакцию фиксации и слежения или счет пальцев — в 7,5 %, различали движение руки у лица — 10 %, светоощущеиие или различимый ЗВП в ответ па вспышку — в 70 %.
Мы наблюдали 7 пациентов с семейной экссудативной витреоретинопатией в возрасте 5—62 лет. Острота зрения при I стадии заболевания составляла 1,0 (3 глаза), при II стадии — 0,02-0,9 (9 глаз), при III стадии — 0 - светоохцущение с правильной проекцией (2 глаза).
Флюоресцентная ангиография. нос исследование позволяет выявить ранние нарушения в виде аваскулярных или неперфузируемых зон (см. рис. 9.20, б), аномально вытянутых и избыточно ветвящихся сосудов, артериовенозных шунтов и телеангиэктазий даже при бессимптомном течении семейной экссудативной витреоретинопатии. В I—11 стациях болезни отмечают просачивание флюоресцеина через стенки расширенных ретинальнь1Х сосудов и аргериовенозных шунтов, накопление и повышенную проницаемость контрастного вещества в областях фиброваскулярной пролиферации и неоваскуляризации. Вдоль границы раздела васкулярной и ненерфузируемой сетчатки эти изменения приобретают конфигурацию, образно называемую «морским веером» (см. рис. 9.20, б).
W. hiMiiuM и cicim (1981) при офтальмоскопическом обследовании IS больных из трех семей не обнаружили изменений у 73 % ИЗ них. В то же время при флюоресцентной ангиографии у этих пациентов были выявлены неперфузируемыезоны на крайней периферии.
Электро физиологические исследования. ЭРГ у пациентов с I стадией заболевания нормальная или субнормальная. Во II и III стадиях болезни ЭРГ значительно снижена или отсутствует.
Дифференциальная диагностика. Семейную JM.W lanc?iyia фсогк: гннопатию необходимо дифференцировать от первичного персистирующего гиперпластического стекловидного тела, ретинопатия недоношенных, болезни Вагнера, Х-сцепленного ювенильного ретиношизиса, болезни Коатса, синдрома Блоха—Сульцбергера (синдром недержания пигмента), болезни Норри, серповидно-клеточной ретинопатии, ретинобластомы, болезни Илза, периферического увеита.
Первичное персистирующее гиперпластическое стекловидное тело (ППГСТ) — редко наблюдаемая спорадическая врожденная аномалия, обусловленная персистенцией и вторичным фиброзом примитивной гиаловдной сосудистой системы. У пациентов с ППГСТ фиброваскулярный тяж отходит от диска зрительного нерва и прикрепляется к задней капсуле хрусталика (рис. 9.24),
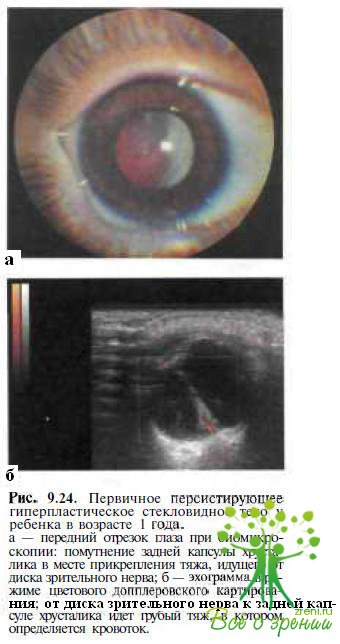
приводя к ее помутнению в месте контакта. Нередко у больных с ППГСТ уменьшен диаметр роговицы. В подавляющем большинстве случаев аномалия односторонняя. Как правило, при отсутствии осложнений, например тракционной отслойки сетчатки, ЭРГ у пациентов с ППГСТ нормальная. Сочетания с системной патологией у пациентов с ГШ ГСП не обнаружено.
А. Chang-Godinich и соавт. (1999) описали необычный случай семейной экссудативной витреоретинопатии у доношенной новорожденной девочки. При осмотре ребенка в возрасте 6 дней авторы обнаружили микрофтальм правого глаза, сочетавшийся с фиброзом стекловидного тела. При исследовании левого глаза патологических изменений не отмечено. На основании по леченных данных был установлен диагноз ППГСТ. Через 3 мес при офтальмоскопии левого глаза были выявлены изменения ретинальных сосудов сетчатки и фиброваскулярная ткань на крайней периферии, что послужило основанием для пересмотра диагноза и проведения лазерной коагуляции аваскулярной зоны сетчатки. В дальнейшем, несмотря на проведенное лечение, у ребенка развилась отслойка сетчатки слева, Изменений в состоянии правого глаза не произошло, Предпринятая дополнительная криотерапия привела к регрессу сосудистых нарушений, резорбции экссудата и прилеганию сетчатки.
Таким образом, семейная экссудативная витреоретинопатия может манифестировать в неонатальном периоде, при этом поражения обоих глаз возникают со значительной разницей во времени, различаясь по тяжести и характеру течения. У новорожденных с односторонним фиброзом стекловидного тела необходимо тщательное исследование парного глаза в динамике для исключения спорадических случаев экссудативной витреоретинопатии.
Ретинопатия недоношенных. Затруднения могут возникнуть при дифференциальной диагностике с рубцовой фазой ретинопатии недоношенных в III—IV стадии. У детей с ретинопатией недоношенных часто выявляют гетеротопию макулы, обусловленную образованием серповидной складки сетчатки, идущей от диска зрительного нерва в нижненаружном квадранте (см. рис. 9.14), грубую деформацию диска зрительного нерва (рис. 9 25),
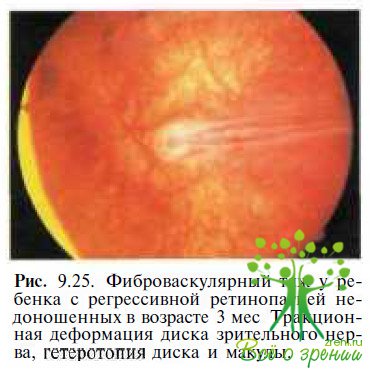
множественные хориоретинальные атрофические очаги, тракционную отслойку сетчатки. ЭРГ у пациентов с ретинопатией недоношенных III—IV стадии обычно отсутствует. Существенное значение для дифференциальной диагностики в этом случае имеют данные анамнеза.
Болезнь Вагнера передается по аутосомно-доминантному типу. Пора¬жаются одинаково часто и мужчины, и женщины. Острота зрения у пациентов долго сохраняется нормальной, отмечается концентрическое сужение поля зрения. Характерна высокая миопия. Макула обычно остается интактной, на периферии определяется ретиношизис. В стекловидном теле выявляют белые васкулнризированные мембраны, локализующиеся преимущественно в экваториальной зоне. После 10 лет развивается катаракта.
Болеть Гольдманна—Фавра — редко наблюдаемое заболевание с аутосомно-рецессивным типом наследования. В отличие от семейной экссудативной витреоретинопатии, которая может манифестировать уже в первые дни жизни, у пациентов с болезнью Гольдманна—Фавра жалобы на плохое зрение в сумерках появляются обычно в возрасте 5—7 лет, после чего постепенно снижается острота зрения и возникают нарушения в поле зрения. По мере прогрессирозаннн заболевания выявляют фибриллярную дегенерацию и лентоподобные мембраны в стекловидном теле, периферический ретиношизис, кистевидную макулопатию (рис. 9.26)
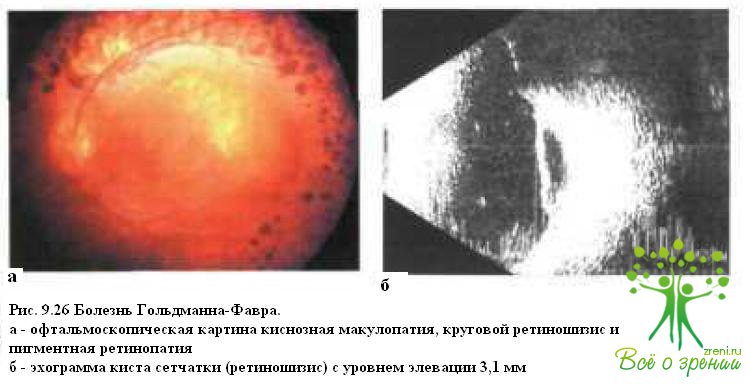
и отложения пигмента в виде костных телец или полиморфных глыбок темно-коричневого цвета (см. рис. 9.18 и 9.19), сужение сосудов в области «клинически нормальной сетчатки» и полную их облитерацию в области ретиношизиса. В возрасте 20—30 лет у большинства больных формируется задняя чашеобразная катаракта. При флюоресцентной ангиографии на периферии определяют обширные неперфузируемые зоны. ЭРГ значительно снижена или отсутствует.
Х-сцепленный ювенильный ретиношизис. У детей раннего возраста с буллезным Х-сцепленным врожденным ретиношизисом может накапливаться интра- и субретинальный твердый экссудат (см. рис. 9.8; рис. 9.27),
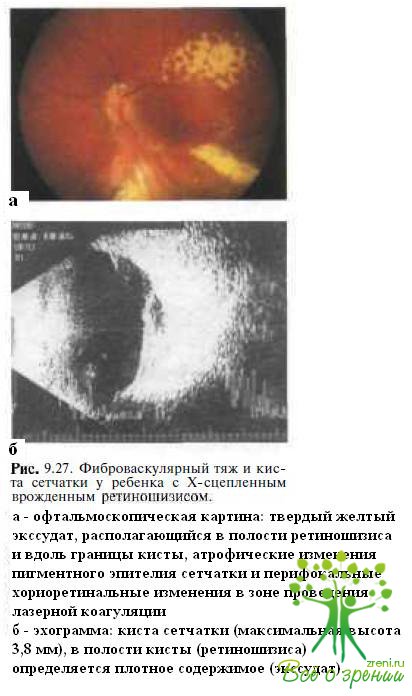
развиваются периферические полиморфные сосудистые изменения в виде периваскулярных муфт, неоваскуляризации, микроаневризм и облитерированных сосудов. Иногда течение заболевания осложняется гемофтальмом, формированием преретинальных фиброглиальных масс, развитием экссудативной отслойки сетчатки. При эхографии в таких случаях выявляют отслойку сетчатки с плотным субретинальным содержимым, что также может вводить офтальмологов в заблуждение. Описаны казуистические случаи развития неоваскулярной глаукомы у детей раннего возраста с Х-сцепленным ювенильным ретиношизисом и тотальной отслойкой сетчатки.
Преодолеть возникающие в ряде случаев диагностические трудности помогают данные семейного анамнеза и результаты офтальмоскопии: наличие кистозной макулопатии, отсутствие гетеротопии макулы и серповидной складки сетчатки у большинства пациентов с врожденным ретиношизисом. Важное значение имеют также результаты флюоресцентной ангаографии (наличие неперфузируемых зон, сосудистые нарушения в виде морского веера на границе аваскулярной и васкулярной областей сетчатки у больных с семейной экссудативной витреоретинопатией)
Болезнь Коатса. Необходимость в дифференциальной диагностике возникает главным образом у пациентов с болезнью Коатса в далеко зашедшей или терминальной стадиях. В отличие от семейной экссудативной витреоретинопатии болезнь Коатса — одностороннее спорадическое заболевание, характеризующееся наличием полиморфных сосудистых аномалий (микро- и макроаневризм, телеангиэктазий, артериовенозных шунтов) и массивной интра- и субретинальной экссудацией, приводящей к развитию экссудативной отслойки сетчатки (рис. 9.28).
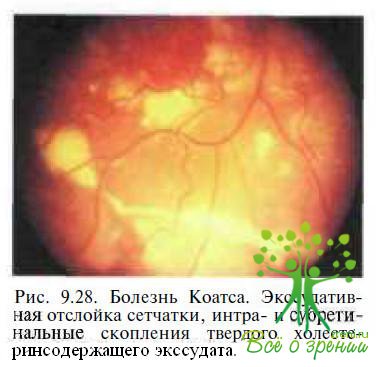
Изменений в стекловидном теле не бывает, за исключением вторичной воспалительной реакции и задней гиалоидной отслойки, наблюдаемой у пациентов в развитой и далеко зашедшей стадиях болезни.
Ретинобластома. Трудности в диагностике возникают только у больных с тотальной или субтотальной отслойкой сетчатки в терминальной стадии заболевания (рис. 9.29).
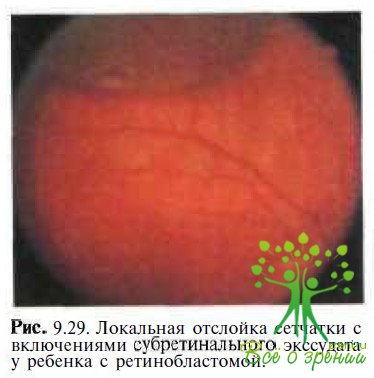
В таких случаях наиболее весомым аргументом, свидетельствующим о наличии ретино- бластомы, является обнаружение эффекта «плюс ткани» и/или кальцификатов при КТ или эхографии (рис. 9.30).

Болезнь Илза — идиопатическая окклюзионная васкулопатия, характеризующаяся формированием обширных неперфузируемых зон в периферических отделах сетчатки, экстраретинальной неоваскуляризацией (у 84 %), рецидивирующими гемофтальмами (у 54 %) и витреоретинальной пролиферацией. Болезнь Илза чаще развивается у мужчин молодого возраста (3 : 1). У 17—48 % больных витреоретинальные изменения сочетаются с вестибулокохлеарными дисфункциями. Заболевание носит эндемичный характер — болезнь Илза распространена на территории Индии и Ближнего Востока. Первые симптомы заболевания появляются в возрасте 12—30 лет, макула длительно сохраняется интактной. Острота зрения в отдаленном периоде у больных превышает 0,5. При ФАГ отмечают признаки венозной окклюзии — дилатацию и извитость сосудов, их окклюзию, наличие микроаневризм и неперфузируемых зон, задержку контрастирования сосудистого русла и ремоделирование сосудов.
Синдром Блоха—Сульцбергера. Заболевание обычно летально для лиц мужского пола, поэтому девочки составляют 97 % живущих с данной патологией. Изменения глаз отмечают у 35—77 % больных. При офтальмоскопии могут выявляться аваскулярные зоны (лучше визуализируются при ФАГ) с неоваскуляризацией или без нее, складки сетчатки, распространяющиеся от диска зрительного нерва в наружной половине, гипер- или гипопигментация, височная гетеротопия диска и макулы, атрофия зрительного нерва. Отслойку сетчатки и ретролентальную пролиферацию отмечают у 23 % больных. У 17 % пациентов развиваются незначительные помутнения роговицы.
Дифференциальный диагноз основывается на результатах дерматологического, стоматологического и неврологического обследования. В отличие от семейной экссудативной витреоретинопатии синдром Блоха—Сульцбергера — мультисистемное заболевание, так как приблизительно у 80 % пациентов отмечаются экстраокулярные нарушения: пигментные изменения кожи (в старшем возрасте они могут быть уже не так заметны), алопеция, патология зубов, неврологическая симптоматика. Заболевание развивается в первые дни или месяцы после рождения. Для острой фазы при поражениях кожи характерно появление на туловище и конечностях эритематозно-везикулезной сыпи, Во II стадии наблюдаются папулезные высыпания, гиперкератические образования, Через несколько месяцев эти высыпания регрессируют. Параллельно появдяются пигментные пятна темно-коричневого цвета, имеющие причудливые очертания, Они могут существовать многие годы, постепенно исчезая ко 2—3-му десятилетию жизни. В грудном возрасте у пациентов отмечают лейкоцитоз и эозинофилию (до 50 %).
Болезнь Норри. Диагностические затруднения могут возникать при дифференциальной диагностике с Х-сцепленной формой семейной экссудативной витреоретинопатии, Двусторонняя отслойка сетчатки или ретролентальные массы, как правило, выявляются у детей с болезнью Норри уже при рождении, Кроме того, для болезни Норри характерны системные отклонения: тугоухость и задержка психического развития, которая при соответствующей психологической реабилитации может отсутствовать, Иногда диагноз можно установить только на основании гистологических исследований — для болезни Норри характерны признаки дисплазии сетчатки.
Токсокароз. Токсокарозный увеит проявляется выраженной воспалительной реакцией в стекловидном теле и наличием бело-серых масс в заднем полюсе или на периферии сетчатки (рис. 9.31). В дальнейшем формируются грубые витреоретинальные тяжи и мембраны. Как в острой стадии заболевания, так и в период ремиссии возможно развитие отслойки сетчатки.
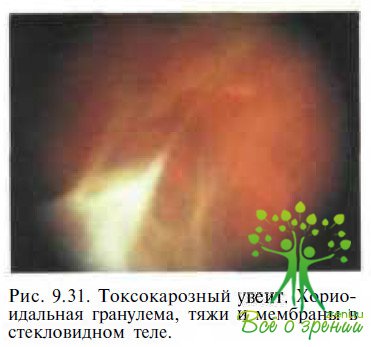
В отличие от семейной экссудативной витреоретинопатии при токсокарозном увейте поражается, как правило, один глаз, а витреальные и экссудативные изменения менее выражены. Кроме того, у 60—80 % больных с токсокарозом в острой фазе заболевания отмечают различные соматические расстройства: лихорадку, уртикарные высыпания на коже, кашель и хрипы в легких, гепатоспленомегалию, судороги, эозинофилию крови. Позднее при рентгенологическом исследовании органов рудном клетки в легких выявляют рассеянные инфильтратнвиые теки. У 45 % пациентов с токсокарозом глаз при иммунологических исследованиях (ферментоподобная иммуносорбентная проба с использованием яиц паразита в качестве антигена) титр антител превышает 1 : 32. Косвенным подтвер¬ждением диагноза у ребенка, у которого заподозрен токсокарозный увеит, является выраженный терапевтический эффект при назначении кортикостеровдов и диэтилкарбамазина.
Лечение. Важными аспектами терапии являются ранняя идентификация начальных изменений сетчатки у детей с семейной экссудативной витреоретинопатией в пораженных семьях, лечение амблиопии при асимметричном поражении. Детей в возрасте до 7 лет с I или начальными признаками II стадии семейной экссудативной витреоретинопатии необходимо ежемесячно обследовать в связи с опасностью быстрого бессимптомного прогрессирования заболевания. В более старшем возрасте пациенты могут отметить признаки прогрессирования витреоретинальных изменений на основании появления метаморфопсий, снижения остроты зрения или изменений в поле зрения, поэтому интервалы между их визитами к офтальмологу могут составлять 4—6 мес. Пациенты в течение всей жизни могут ожидать развития осложнений. W.E. Benson (1995), анализируя результаты длительного наблюдения 37 больных с семейной экссудативной витреоретинопатией, сообщил о развитии отслойки сетчатки у 5 из них через 6—17 лет после клинической стабилизации заболевания.
В случае прогрессирования болезни, а также при фиброваскулярной пролиферации и/или неоваскуляризации показаны криотерапия или лазерная коагуляция аваскулярных зон, новообразованных сосудов и областей пролиферации. К сожалению, криотерапия и/или лазерная коагуляция сетчатки не во всех случаях позволяют достичь стабилизации заболевания, а иногда инициируют формирование разрывов сетчатки и развитие отслойки. Учитывая возможность самопроизвольной регрессии, а также высокий риск развития осложнений, обусловленных проведением криотерапии и/или лазерной коагуляции сетчатки, при наличии у пациентов небольших веточек неоваскуляризации можно ограничиться наблюдением и проведением медикаментозного лечения, направленного на улучшение микроциркуляции и метаболизма сетчатки.
Отслойка сетчатки у большинства пациентов развивается до 10-летнего возраста. В этих случаях показана витрэктомия в сочетании с экстрасклеральным пломбированием и иссечением эпиретинальных мембран. T. Ikeda и соавт. (1999) отмечают, что из-за выраженной витреоретинальной адгезии при иссечении эпиретинальных мембран часто (в 22 случаях из 28) возникают ятрогенные разрывы сетчатки. Хирургическое лечение отслойки сетчатки у пациентов с семейной экссудативной витреоретинопатией позволяет достичь не только прилегания сетчатки, но и сохранить предметную остроту зрения у 35—67 % из них даже при тракционной отслойке макулы.
Необходимо генетическое консультирование членов пораженных семей с бессимптомным течением семейной экссудативной витреоретинопатии, так как у их детей заболевание может протекать в более тяжелой форме.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0