Врожденный амавроз Лебера и М.Mосин


Описание
Впервые заболевание, обозначаемое в литературе эпонимом «врожденный амавроз Лебера», было описано Т. Leber в 1867 г. Характерные признаки заболевания, указанные Т. Leber, — врожденная слепота, маятниковый нистагм и развитие пигментных нарушений сетчатки в более старшем возрасте.В настоящее время принято считать, что врожденный амавроз Лебера — гетерогенная группа врожденных заболеваний, составляющих около 5 % случаев от общего числа наследственных ретинальных дистрофий.
Диагностика врожденного амавроза Лебера основывается на 3 критериях:
- выраженные нарушения зрительных функций в раннем возрасте;
- отсутствие ЭРГ или ее значительное снижение;
- наличие пигментной ретинопатии в первые месяцы жизни или в более старшем возрасте.
Генетические исследования. Полиморфизм клинических проявлений при патологических состояниях, сгруппированных под названием «врожденный амавроз Лебера», не оставляет никаких сомнений относительно того, что речь идет о ряде мутаций, связанных с разными генами. В подавляющем большинстве случаев отмечается аутосомно-рецессивный тип наследования, хотя описаны несколько семей с аутосомно-доминантным типом передачи.
К 2002 г. были верифицированы 6 разных генов, мутации в которых приводят к развитию врожденного амавроза Лебера. Эти гены кодируют синтез продуктов с различными функциями: регулятор эмбрионального развития сетчатки (CRX), фототрансдуктор (GUCY2D), регулятор структуры фоторецепторов (CRB1), транспортные белки (AIPLI, RPGRIP1), протектор метаболизма витамина A (RPE65).
Один из генов, ответственных за развитие врожденного амавроза Лебера, был локализован в 17-й хромосоме I. Perrault и соавт. (1996). Ген располагается в локусе 17р13.1 и получил название LСAl Ген LСAl выявили у членов лишь 8 из 15 обследованных североафриканских семей, в которых были обнаружены больные с врожденным амаврозом Лебера. Это также свидетельствовало о генетической гетерогенности заболевания. Исследовав 4 пробандов из других североафриканских семей, в которых заболевание не было обусловлено мутациями гена LCA1, авторы обнаружили две миссенсмутации (F589S) и две мутации, приводящие к сдвигу рамки считывания (nt 460 del С, nt 693 del С), в гене ретинальной гуанилатциклазы (RETGC, GDB symbol GUC2D). На основании этих фактов авторы предположили, что ген LCAI ответствен за нарушение продукции циклической гуанозинмо-нофосфатазы (цГМФ) в сетчатке и, следовательно, за прекращение работы ретинальных цГМФ-регулируемых катионных каналов.
Приблизительно у 7—9 % пациентов с врожденным амаврозом Лебера, передающимся по аутосомно-доминантному типу, отмечаются мутации в гене AIPL1 (арилгидрокарбон рецепторин-терактивирующий белковоподобный 1). Ген AIPLI кодирует продукцию белка, состоящего из 384 аминокислот, функция которого пока неизвестна. Возможно, ген AIPL1 ответствен за регуляцию клеточного цикла фоторецепторов. Его мутации приводят к гибели фоторецепторов в период развития.
В ряде случаев развитие врожденного амавроза Лебера связывают с гомозиготными мутациями в гене КРЕ65. В модели на животных было установлено, что мутации в гене KPE6S опосредуют дегенерацию фоторецепторов. Изменения гена RPE6S приводят к накоплению эфиров полностью — трансретинола в пигментном эпителии сетчатки, обусловливая дефицит 11-цисретинола и в итоге родопсина.
Гистологические исследования. В литературе имеются единичные сообщения о гистологических исследованиях при врожденном амаврозе Лебера. Их авторы отмечают у больных отсутствие в сетчатке слоя фоторецепторов, вместо которого определяется слой кубовидных клеток. В периферических областях сетчатки выявляют зоны миграции пигмента, глиоза, а также выраженную деструкцию слоя пигментного эпителия сетчатки и мембраны Бруха.
Клинические проявления. Как правило, родители замечают у ребенка отсутствие фиксации взгляда или реакции на свет уже в первые 3—4 мес жизни. У всех пациентов с врожденным амаврозом Лебера обращает на себя внимание атрофия периорбитальной клетчатки. Родители каждого пятого пациента отмечают, что их ребенок тычет себе в глаза пальцами или трет их. Этот характерный для пациентов с амаврозом Лебера феномен был впервые описан в 1947 г. и называется по имени автора окулопальцевым симптомом Францескетти. У 1/3 детей отмечают светобоязнь, в 53—75 % случаев — нистагм или блуждающие движения глаз.
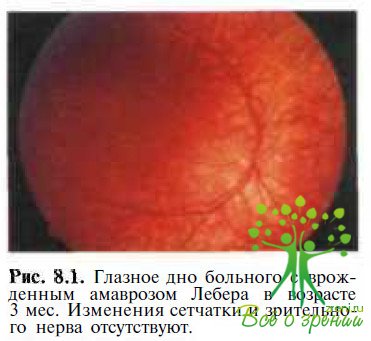
При офтальмоскопии глазное дно выглядит нормальным примерно у половины пациентов в течение первого года жизни (рис. 8.1).
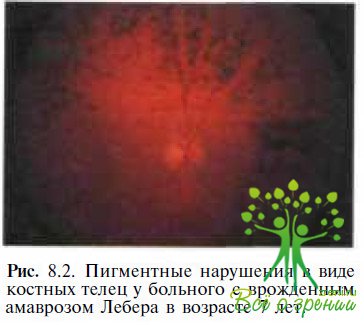
С течением времени появляются изменения в сетчатке: гранулярные пигментные образования (рис. 8.2), «соль с перцем», бело- точечный ретинит, «мраморное» глазное дно, хориоретинальная атрофия или колобомоподобные поражения.
В возрасте 2—3 лет у части пациентов, не имевших ранее изменений на глазном дне, появляются побледнение диска зрительного нерва и сужение ретинальных артериол. Атрофию зрительного нерва фиксируют у 10—70 % пациентов с амаврозом Лебера. Имеются сообщения о других аномалиях диска зрительного нерва у детей с врожденным амаврозом Лебера, среди которых наиболее часто (примерно у 3 %) встречается псевдозастой.
Столь выраженные различия в частоте выявления атрофии зрительного нерва у пациентов с врожденным амаврозом Лебера обусловлены отличиями в диагностических критериях, которые использованы авторами для идентификации болезни у детей раннего возраста. Учитывая данное обстоятельство, а также широкий спектр заболеваний, при которых ЭРГ в первые месяцы жизни не регистрируется или значительно редуцирована, нельзя исключить вероятности того, что авторами некоторых из упомянутых выше сообщений врожденный амавроз Лебера мог быть ошибочно диагностирован у детей с атрофией зрительного нерва, возникшей на фоне системных поражений.
В более старшем возрасте возможно развитие кератоглобуса, кератоконуса и катаракты.
Большинство пациентов с врожденным амаврозом Лебера (до 86 %) имеют высокую гиперметропию, в то же время описана подгруппа детей с амаврозом Лебера, имеющих миопическую рефракцию.
R.K, Koenekoop и соавт. (2002) сообщали о пробанде и его матери с врожденным амаврозом Лебера, у которых была обнаружена гетерозиготная мутация CRX, A177Deltalbp (529delG). При длительном наблюдении авторы отметили самопроизвольное постепенное незначительное улучшение зрения у мальчика. В пятимесячном возрасте у ребенка констатировали отсутствие фиксации, нистагм, сужение артериол, парадоксальную реакцию зрачков на свет и неразличимую ЭРГ. В возрасте 6 лет острота его зрения составляла 20/900, а в 11 лет — уже 20/150. В возрасте 10 и 11 лет у пациента регистрировали фотопическую ЭРГ, в которой определялись как а-, так и Ь-волны. При периметрии объектом V/4 в возрасте 9 и 10 лет у мальчика удалось установить периферические границы поля зрения в пределах 60°, а позднее были обнаружены новые островки в 20° в нижней половине поля зрения при использовании объекта Ш/4. У 47-летней матери пробанда отмечались пигментные изменения сетчатки, острота зрения составляла «движение руки у лица», а ЭРГ и поле зрения были неопределяемыми.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
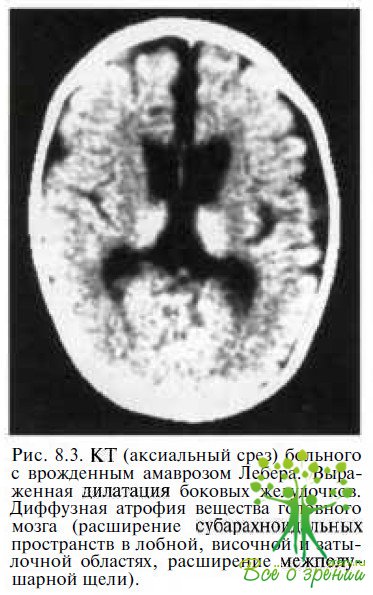
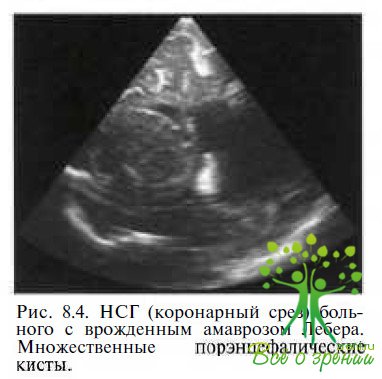
Сочетание с системными заболеваниями. Неврологическую симптоматику обнаруживают в 17—37 % случаев заболевания, в частности у детей — судорожный синдром. У каждого второго ребенка с врожденным амаврозом Лебера при нейросонографии или радиологических исследованиях выявляют изменения в головном мозге: вентрикуломегалию различной степени выраженности и/или диффузную атрофию мозга (рис. 8.3),
порэнцефалию (рис. 8.4),
гипоплазию червя мозжечка (рис. 8.5).
У 15-50% детей диагностируют задержку психомоторного развития. Между тем, по мнению A.Steinberg и соавт. (1992), задержка психомоторного развития у детей с врожденным амаврозом Лебера встречается не чаще, чем у детей с врожденной слепотой другой этиологии, и когнитивные способности детей с врожденным амаврозом Лебера не ниже, чем у других детей, слепых с момента рождения. Кроме того, у детей с врожденной слепотой независимо от ее этиологии значительно чаще отмечают мышечную гипотонию и снижение двигательной активности, чем у здоровых младенцев того же возраста. Таким образом, встречающаяся у части детей с врожденным амаврозом Лебера задержка психомоторного развития во многом обусловлена сенсорной депривацией в сенситивный период в первые месяцы жизни. Следовательно, ранняя стимуляция развития, включающая психологическую реабилитацию, необходима для более полной социальной адаптации пациентов с врожденным амаврозом Лебера.
Более редкие системные проявления, сочетающиеся с амаврозом Лебера, включают глухоту, кардиомиопатию, ювенильный нефронофтиз и остеопороз. Впрочем, сообщение V. Godel и соавт. (1978) о наблюдаемом ими случае сочетания врожденного амавроза Лебера с ювенильным нефронофтизом представляет собой не что иное, как описание редко встречающегося синдрома Сениора— Боичиса (см. «Дифференциальная диагностика»), который пятью годами ранее подробно охарактеризовали в литературе H. Boichis и соавт. (1973) и несколько позже В. Stanescu и соавт. (1976). В 1980 г. V. Godel и соавт. сообщили еще об одном случае сочетания ювенильного нефронофтиза с секторальным пигментным ретинитом у 18-летней девушки. В ходе психофизических и электрофизиологических исследований у пациентки были обнаружены нормальная острота зрения, верхненосовая квадрантопсия, соответствующая зоне гиперпигментации сетчатки, и субнормальная ЭРГ. Эти факты еще раз указывают на то, что под диагнозом «амавроз Лебера» объединяют гетерогенную группу врожденных состояний, патогенез и клинические особенности которых изучены недостаточно. В связи с этим во избежание диагностических ошибок необходимо дифференцировать амавроз Лебера от других мультисистемных аномалий, одним из симптомов которых является пигментный ретинит.
Электрофизиологические исследования. Проведение электрофизиологических исследований является неотъемлемой частью обследования детей раннего возраста с нарушенным зрением. Регистрация ЭРГ и ЗВП в отпет на вспышку и/или паттерны позволяет не только оценить степень тяжести функциональных нарушений, по и получить информацию, необходимую для дифференциальной диагностики. Одним из наиболее важных диагностических критериев амавроза Лебера является отсутствие ЭРГ (рис. 8.6).
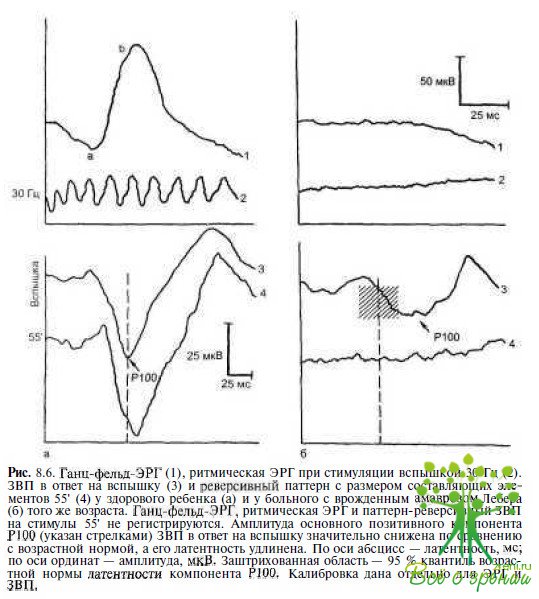
Л.С. Орловская (1984), обследовав 112 детей с амаврозом Лебера, обнаружила отсутствие ЭРГ у 93 % пациентов, у остальных больных амплитуда ЭРГ была субнормальной. Резко сниженные по амплитуде ЗВП в ответ на вспышку света отмечают лишь у половины пациентов. ЗВП в ответ на реверсивные паттерны не регистрируются практически во всех случаях (см. рис. 8.6).
Несмотря на выраженные нарушения ЭРГ, ее значительное снижение или отсутствие, установленные в ходе первого обследования в неонатальном периоде, нельзя безоговорочно трактовать как свидетельство в пользу амавроза Лебера, даже при наличии изменений на глазном дне. Существует множество системных заболеваний, сопровождающихся развитием ретинопатии, при которых у младенцев ЭРГ не регистрируется или значительно редуцирована (см. «Дифференциальная диагностика»). При некоторых заболеваниях из этой группы больных электроретинограмму, отсутствующую в неонатальном периоде, удается записать в более старшем возрасте. Следовательно, в сомнительных ситуациях для диагностики врожденного амавроза Лебера необходимы дополнительные исследования для уточнения соматического статуса пациентов, а в ряде случаев — длительное наблюдение за детьми и повторное электрофизиологическое обследование в более старшем возрасте (обычно через 3—6 мес).
Дифференциальная диагностика. В настоящее время врожденный амавроз Лебера рассматривают как гетерогенную группу заболеваний, характеризующихся врожденной потерей зрения, отсутствием ЭРГ и ретинальными пигментными изменениями, возни— кающими в более старшем возрасте. Однако существует множество как глазных, так и системных заболеваний, имеющих симптоматику, сходную с врожденным амаврозом Лебера, и вследствие этого нередко ошибочно трактуемых. Установление правильного диагноза в раннем возрасте и своевременная терапия при некоторых из этих патологических состояний, например при инфантильном злокачественном остеопетрозе или инфантильной форме болезни накопления фитиновой кислоты, позволяет не только сохранить пациентам жизнь, но и предотвратить неминуемую слепоту.
При подозрении у младенцев врожденного амавроза Лебера необходимо проводить дифференциальную диагностику со следующими заболеваниями:
- врожденная стационарная ночная слепота;
- церебральные зрительные поражения и корковая слепота;
- врожденная атрофия зрительного нерва;
- задержка зрительного созревания;
- врожденная глазодвигательная апраксия;
- болезньБаттена (Batten);
- болезнь Барде—Бидля (Bardet— Biedl);
- болезнь Тея-Сакса (Тау—Sachs);.
- инфантильный злокачественный остеопетроз (болезнь Albers—Schonberg);
- болезнь Жобера (Joubert);
- синдромы Сениора—Локена (Seni¬or— Loken) и Сениора—Боичиса (Senior— Boichis);
- синдром Альстрема (Alstrom);
- синдром Рефсума (Refsum) и инфантильная форма болезни накопления фитиновой кислоты;
- I тип синдрома Ушера;
- асфиксическая дисплазия грудной клетки Жене (Jeune);
- оливомосто-мозжечковая атрофия (ОММА), тип II.
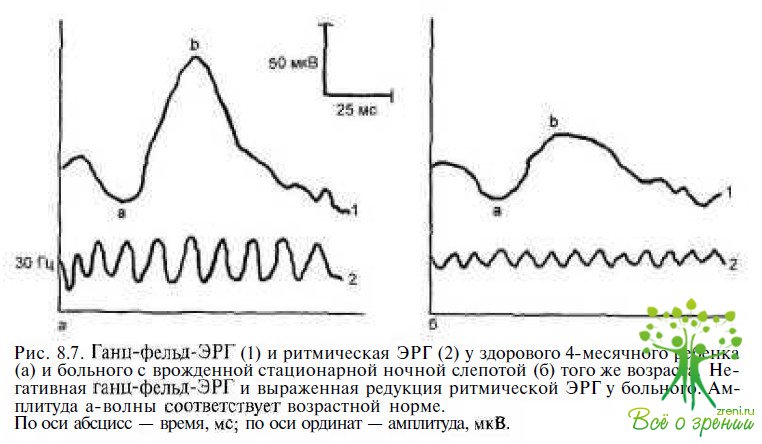
Врожденная стационарная ночная слепота (ВСНС) характеризуется нарушениями скотопического зрения. Острота зрения у этих пациентов в старшем возрасте составляет 0,1—1,0, поэтому значительных поведенческих отклонений в неональном периоде, свидетельствующих о зрительных расстройствах, у них не отмечают. Изменений на глазном дне при ВСНС, в отличие от амайроза Лебера, как правило, не бывает, за исключением некоторых редких аутосомно-рецессивных форм ВСНС (белоточечная абиотрофия, болезнь Огуши). У пациентов с ВСНС нет нистагма и косоглазия. Большинство пациентов с ВСНС в отличие от детей с врожденным амаврозом Лебера имеют миопическую рефракцию. Ключевым тестом для дифференциальной диагностики ВСНС и врожденного амавроза Лебера является ЭРГ. При Х-сцепленной и аутосомно-рецессивной формах ВСНС регистрируется негативная ЭРГ (рис. 8.7), характеризующаяся снижением амплитуды b-волны в скотопических условиях, так называемый тип Шуберта— Борншейна. При аутосомно-доминантнои форме ВСНС встречается более редкий тип электрофизиологических отклонений (тип Риггса), когда скотопическая ЭРГ отсутствует, но фиксируется субнормальная фотопическая ЭРГ.
Церебральные зрительные поражения — полиэтиологический синдром, развивающийся в результате патологических изменений геникулостриарных путей и сопровождающийся стойкими нарушениями зрительных функций. Наиболее частыми причинами церебральных зрительных поражений у младенцев являются пренйтапьные гипоксически-ишемические или геморрагические поражения ЦНС, внутриутробные нейроинфекции, токсико-дисметаболические расстройства и гидроцефалия. Клиническая картина церебральных зрительных поражений характеризуется отсутствием оптокинетического нистагма, нормальной офтальмоскопической картиной и сохранными зрачковыми реакциями.
Дифференциальная диагностика в первые месяцы жизни основывается на результатах электрофизиологических исследований. В отличие от детей с врожденным амаврозом Лебера у пациентов с церебральными зрительными поражениями — нормальная ЭРГ. Амплитуда основного позитивного компонента Р100 ЗВП в ответ на вспышку, как правило, снижена, а латентность удлинена, хотя в некоторых случаях эти параметры могут быть нормальными. В тех случаях, когда у младенцев подозревают церебральные зрительные поражения, а электрофизиологическое обследование подтверждает наличие зрительных нарушений, необходимы нейровизуальные исследования, включающие ней- росонографию и/ил и радиологические методы (МРТ и КТ головного мозга) для уточнения генеза и локализации патологического процесса.
Врожденная атрофия зрительного нерва — поражение зрительного нерва, обусловленное дегенерацией его аксонов и сопровождающееся снижением зрительных функций. У грудных детей атрофия зрительного нерва чаще является следствием перенесенных внутриутробных или постнатальных нейроинфекций, гидроцефалии, наследственных метаболических нарушений. Учитывая возрастные особенности глазного дна в первые месяцы жизни, диагностировать атрофию зрительного нерва у детей грудного возраста, основываясь лишь на данных офтальмоскопии, достаточно сложно.
Кроме того, в некоторых случаях при отсутствии данных полного электрофизиологического обследования (включая различные модификации ЭРГ и ЗВП) обнаруженная у ребенка атрофия зрительного нерва может быть ошибочно интерпретирована как единственная причина снижения зрения. Между тем атрофия зрительного нерва встречается у 10—70 % пациентов с амаврозом Лебера. Чтобы дифференцировать врожденный амавроз Лебера от атрофии зрительного нерва у детей раннего возраста, необходимы электрофизиологические исследования. У пациентов с атрофией зрительного нерва практически во всех случаях регистрируют нормальную ЭРГ, а ЗВП характеризуются нарушенной конфигурацией, снижением амплитуды и удлинением латентности основного позитивного компонента Р100.
Задержка зрительного созревания — идиопатическое транзиторное отсутствие зрительных реакций у грудных детей. При задержке зрительного созревания в ходе нейроофтальмологического обследования не обнаруживается каких-либо отклонений от возрастной нормы. Родители в таких случаях отмечают у ребенка отсутствие фиксации взора и реакции на или знакомые лица в возрасте 2—3 Нередко задержка зрительного созревания сочетанием л моторном развитии. В отличие от больных с врожденным амаврозом Лебера у детей с задержкой зрительного созревания нет нистагма и изменений на глазном дне, а параметры ЭРГ и ЗВП в ответ на вспышку не отличаются от возрастной нормы.
ЗВП в ответ на реверсию паттернов могут быть незначительно снижены по амплитуде. При НСГ и/или нейрорадиологических исследованиях не вываляется изменений вещества головного мозга в проекции зрительных путей. Как показывает наш опыт, при повторном обследовании через 3—6 мес у таких детей поведенческие зрительные реакции и параметры патгерн-ЗВП соответствуют возрасту. В большинстве случаев у таких детей позднее развивается нормальное зрение.
Врожденная глазодвигательная спраксия — гетерогенная группа патологических состояний, характеризующихся нарушением движений глаз при относительной сохранности зрительных функций. Взрослые пациенты с апраксией при смене точки фиксации компенсируют этот дефект поворотом головы. У детей первых месяцев жизни контроль за движениями головы еще не развит, поэтому при их осмотре возникает ошибочное впечатление о возможных зрительных расстройствах.
Для дифференциальной диагностики необходимо провести электрофизиологические исследования: в отличие от выраженного снижения амплитуды биопотенциалов сетчатки и зрительной проекционной области коры головного мозга, наблюдаемых при врожденном амаврозе Лебера, амплитудные показатели ЭРГ и ЗВП у детей с врожденной глазодвигательной апраксией близки к нормальным.
Болезнь Баттена — заболевание с аутосомно-рецессивным типом наследования, имеющее прогрессирующее течение и характеризующееся зрительными расстройствами, деменцией, судорожными припадками и спастическими пара- или тетрапарезами.
При инфантильной форме (синдром Халтия—Сантавуори) заболевания у младенцев в возрасте 6—8 мес появляются признаки нарушения психомоторного развития, иногда — микроцефалия. В ходе офтальмологического обследования обычно выявляют атрофию зрительного нерва и пигментную ретинопатию. ЭРГ у младенцев с болезнью Баттена не регистрируется. ЗВП отсутствуют или значительно снижены по амплитуде. ЭЭГ к 3-му году жизни становится изоэлектрической. Полная слепота развивается в возрасте от 2 до 3 лет. Пациенты обычно умирают в возрасте 6—7 лет..
При поздней инфантильной форме (синдром Янского—Бильшовского) первыми симптомами болезни являются судороги, которые развиваются в возрасте от 2 до 4 лет. Позднее появляются признаки прогрессирующей мозжечковой атаксии, задержка психомоторного развития. Пациенты, как правило, погибают до 10 лет.
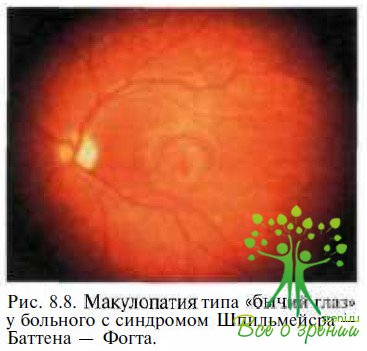
При ювенильной форме заболевания (синдром Шпильмейера—Баттена – Фогта) зрительные нарушения предшествуют неврологическим симптомам. Первым офтальмоскопическим симптомом может быть исчезновение фовеолярного рефлекса и/ ил и макулопатия типа «бычий глаз» (рис. 8.8). Признаки центральных зрительных нарушений появляются в возрасте 3—7 лет. Смерть из-за неврологических осложнений наступает обычно в возрасте 20—40 лет.
Проведение дифференциальной диагностики врожденного амавроза Лебера и болезни Баттена необходимо в тех случаях, когда клиницисты сталкиваются с инфантильной формой заболевания (синдромом Халтия—Сантавуори). В отличие от болезни Баттена, которая манифестирует приблизительно после 6—8 мес жизни у нормально развивавшихся до этого момента детей, клинические симптомы у пациентов с врожденным амаврозом Лебера, свидетельствующие о грубых нарушениях зрения, появляются уже в первые 2—3 мес жизни. В дальнейшем у детей с болезнью Баттена отмечают нарастающую неврологическую симптоматику, формирование изоэлектрической ЭЭГ и прогрессирующую деменцию, что не характерно для пациентов с врожденным амаврозом Лебера.
Помощь в диагностике оказывает морфологическое исследование биоптатов тканей конъюнктивы или слизистой оболочки кишечника. При электронной микроскопии нейронов, перицитов, макрофагов, гладкомышечных клеток, лимфоцитов и клеток эндотелия у пациентов с инфантильной формой болезни Баттена определяются гранулярные осмиофильные депозиты.
Синдром Барде— Бидля — аутосомно-рецессивное мультисистемное заболевание, характеризующееся ожирением, полидактилией, недоразвитием половых органов, задержкой роста, слабоумием и пигментной ретинопатией. Заболевание впервые описано G. Bardel (1920), который сообщил о пациенте с пигментным ретинитом, ожирением и полидактилией. В 1922 г. А. В1есН дополнил два новых признака, характеризующих заболевание: умственная отсталость и гипогенитализм. Ретинальные изменения представлены нерезко выраженными нарушениями пигментного эпителия сетчатки в макуле и незначительной гиперпишентацией в периферических отделах (рис. 8.9). Скотопическая и фотопическая ЭРГ резко снижены.
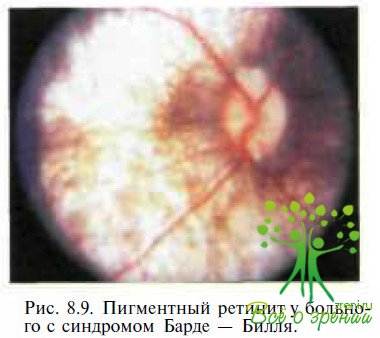
Дифференциальный диагноз с врожденным амаврозом Лебера основывается на очевидных соматических отличиях. От описанной выше инфантильной формы болезни Ватте на синдром Барде—Бидля отличается наличием ожирения, полидактилии, выраженных вегетативных нарушений и отсутствием двигательных расстройств. Важно отметить, что заболевание отличается и от описанного 60 годами ранее синдрома Лоуренса—Муна, при котором пигментный ретинит сочетается с параплегией при отсутствии ожирения и полидактилии.
Болезнь Тея—Сакса — часто встречающийся тип ганглиозидоза, обусловленный отсутствием гексозаминидазы А. У нормально развивающихся до 2—6 мес детей появляются признаки нарушения зрения, нистагм, мышечная гипотония, судороги. Характерным офтальмоскопическим симптомом является вишневый цвет макулы, окруженной желтовато-серым ободком (рис. 8.10). Вишневый цвет макулы обусловлен потерей прозрачности окружающей ее сетчатки из-за накопления лип вдов в ганглиозных клетках.
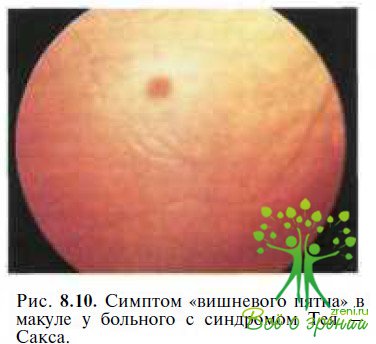
Дифференциальная диагностика при своевременном осмотре основывается на данных офтальмоскопии. В более старшем возрасте в связи с тотальной гибелью ганглиозных клеток сетчатки симптом «вишневого пятна» в макуле исчезает. К концу 1-го года жизни почти у всех пациентов с болезнью Тея—Сакса развивается атрофия зрительного нерва. Подавляющее большинство пациентов слепнут к 18-му месяцу жизни.
Инфактильный злокачественный остеопетроз (болезнь Алъберса-Шонбер- га) — наследственное метаболическое заболевание с аутосомно-рецессивным типом передачи, характеризующееся прогрессирующим разрастанием костной ткани. Манифестирует внутриутробно или в первые месяцы жизни. При отсутствии адекватной терапии пациенты погибают в течение первых 10 лет жизни, как правило, от инфекционных осложнений. Спектр офтальмологических поражений включает нистагм, косоглазие, ограничение подвижности глаз, птоз, атрофию или отек зрительного нерва, пигментную ретинопатию (рис. 8.3 1, а). Резкое снижение зрения обычно ассоциируется с атрофией зрительного нерва, которую выявляют приблизительно у 80 % пациентов.
Как правило, атрофия зрительного нерва развивается в более поздние сроки, а в неонатальном периоде о нарушениях зрения свидетельствуют лишь поведенческие отклонения, нистагм, косоглазие и пигментные изменения сетчатки. ЭРГ у младенцев с инфантильным злокачественным остеопетpoзом снижена, ЗВП на вспышку света либо не регистрируются, либо с трудом выделяются из фоновых шумов. Описанный симптомокомплекс имеет общие черты с врожденным амаврозом Лебера, что требует проведения дифференциальной диагностики в максимально короткие сроки, так как своевременное выявление инфантильного остеопетроза и адекватная терапия, в том числе пересадка костного мозга и декомпрессия зрительного нерва, позволяют в ряде случаев спасти больным жизнь и сохранить остаточное зрение.
В отличие от врожденного амавроза Лебера первыми и доминирующими проявлениями болезни являются неврологические симптомы, включающие чрезмерную возбудимость, параличи черепных нервов, гидроцефалию, дизартрию, задержку психомоторного развития. При проведении КТ и МРТ фиксируют атрофию и/или расширение желудочков головного мозга, а также сужение черепных каналов и отверстий. Кроме того, дифференциальная диагностика основывается на выраженной соматической симптоматике, свойственной инфантильному остеопетрозу. У всех больных отмечают гепатоспленомегалию, анемию, лейкопению, тромбоцитопению, костные деформации и переломы.
Дополнительным отличием врожденного амавроза Лебера и инфантильного остеопетроза является регистрируемый в последнем случае негативный тип ЭРГ: резкое снижение амплитуды b-волны при относительной сохранности параметров а-волны (рис. 8.11, б). У большинства пациентов с врожденным амаврозом Лебера ЭРГ отсутствует или определяется значительное снижение амплитуды всех ее компонентов.

Синдром Жобера. Симптоматика у младенцев с синдромом Жобера напоминает проявления врожденного амав- роза Лебера, что может вводить офтальмологов и педиатров в заблуждение. Заболевание наследуется по аутосомно-рецессивному типу. Как и при врожденном амаврозе Лебера, изменения на глазном дне в младенческом возрасте отсутствуют. В более позднем возрасте выявляют побледнение диска зрительного нерва, сужение артериол и пигментные ретинальные нарушения. Амплитуда ЭРГ обычно снижена. Амплитуда и латентность компонента Р100 ЗВП в ответ на вспышку могут быть изменены незначительно, что является позитивным прогностическим признаком в отношении зрения. Среди других проявлений синдрома Жобера можно выделить отрывистые толчкообразные движения глаз, атаксию, неонатальное тахипноэ, гипоплазию или агенезию червя мозжечка, мальформации ствола мозга, отставание в психическом развитии. Наиболее важными критериями для дифференциальной диагностики являются нейросоматические нарушения, результаты регистрации ЗВП и выявление гипоплазии или агенезии червя мозжечка при МРТ.
Синдром Сениора—Локена — аутосомно-рецессивное состояние, характеризующееся сочетанием пигментной ретинопатии и патологии почек (нефронофтиз). Ген, ответственный за развитие синдрома Сениора—Локена, локализуется на 3-й хромосоме в интервале q2l— q22.
Заболевание в первые месяцы жизни сложно дифференцировать от врожденного амавроза Лебера. ЭРГ у таких детей тоже отсутствует или значительно редуцирована. Единственным отличием синдрома Сениора—Локена от врожденного амавроза Лебера является поликистоз почек, клинические проявления которого в раннем возрасте определяются, главным образом, степенью поражения почек, например их увеличением, хронической или прогрессирующей почечной недостаточностью, гипертензией и повторяющейся гематурией. Дети с синдромом Сениора— Локена при отсутствии своевременного лечения часто погибают в раннем возрасте из-за почечной недостаточности. В связи с этим у всех детей раннего возраста с пигментной ретинопатией и выраженной редукцией ЭРГ необходимо исследовать функции почек и проводить ультразвуковое исследование органов брюшной полости для того, чтобы исключить синдром Сениора—Локена.
Синдром Сениора— Ноичиса — аутосомно-рецессивное заболевание, характеризующееся нефронофтизом, врожденным фиброзом печени, нистагмом и тапеторетинальной дегенерацией. ЭРГ на ранних стадиях болезни субнормальная, но по мере прогрессирования изменений исчезает полностью. В раннем возрасте поражение печени может быть незначительным, но у детей в возрасте 3—4 лет и старше прогрессирующий фиброз околоворотных структур часто приводит к воротной гипертензии, расширению вен пищевода и гепато- и спленомегалии.
У пациентов старшего возраста выделяют дефекты в поле зрения, соответствующие зонам пигментных отложений на сетчатке. Острота зрения в некоторых случаях может оставаться нормальной .
Нарушения функции почек развиваются так же, как и у детей с синдромом Сениора—Локена. Часты анурия или олигурия, макро- или микрогематурия. Диагноз ставят на основании клинической симптоматики, данных электрофизиологических исследований, сонографии и урографии.
Синдром Альстрема — редкое мультисистемное заболевание, характеризующееся атипичной пигментной ретинопатией, ожирением, нейросенсорной глухотой и сахарным диабетом. Сахарный диабет встречается приблизительно у 75 % пациентов с синдромом Альстрема. но, и отличнее от ювенилъного диабета, контролируется медикаментозно или диетой. Офтальмологические нарушения появляются уже в первые месяцы жизни и включают нистагм, светобоязнь и пигментную ретинопатию (изменения по типу «соли с перцем»). В более старшем возрасте возможны атрофия зрительного нерва и задняя субкапсулярная катаракта. Амплитуда ЭРГ и ЗВП у пациентов с синдромом Альстрема резко снижена. Зрительные нарушения прогрессируют. Острота зрения в возрасте 7—Ю лет колеблется у большинства пациентов в пределах от «счета пальцев у лица» до правильной светопроекции.
Дифференциальная диагностика основывается на выраженных системных отклонениях, которые не встречаются у пациентов с амаврозом Лебера.
Инфантильная форма болезни накопления фитиновой кислоты (ИБНФК) и болезнь Рефсума — аутосомно-рецессивные заболевания, обусловленные накоплением в сыворотке крови и тканях фитиновой кислоты. Не исключено, что болезнь Рефсума и ИБНФК представляют собой различные формы одного заболевания.
ИБНФК манифестирует в неонатальном периоде и характеризуется мышечной гипотонией, задержкой психомоторного развития, нейросенсорной глухотой и нарушениями зрения. Офтальмологические проявления включают нистагм, косоглазие, пигментную ретинопатию и атрофию зрительного нерва. При ИБНФК офтальмолог может быть первым, кто распознает заболевание. Раннее лечение ИБНФК, в частности назначение диеты с исключением продуктов, содержащих фитиновую кислоту, позволяет сохранить пациентам жизнь и предметное зрение.
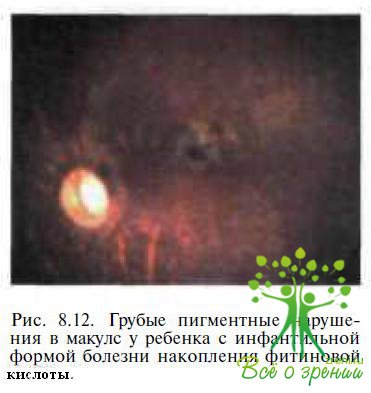
Дифференциальная диагностика основывается на клинических, электрофизиологических и лабораторных критериях. При офтальмоскопии выявляются побледнение диска зрительного нерва и ретинальные пигментные нарушения, локализующиеся, в отличие от таковых при амаврозе Лебера, преимущественно вокруг макулы (рис. 8.12). ЭРГ у детей с ИБНФК субнормальная. Амплитуда основного позитивного компонента Р100 ЗВП снижена, латентность удлинена; в ликворе повышенное содержание белка; в сыворотке крови — высокое содержание фитиновой кислоты, меди, церулоплазмина; в моче — большое количество жирных кислот.
При врожденном амаврозе Лебера редко возникает необходимость дифференциальной диагностики с синдромом Рефсума, так как последний развивается в более старшем возрасте.
Никталопия нередко является одним из первых симптомов заболевания. Пигментная ретинопатия сопровождается прогрессирующим концентрическим сужением поля зрения. Во второй декаде жизни появляются признаки прогрессирующего полиневрита, мозжечковой атаксии, реже — аносмия и снижение слуха, ихтиозоподобные изменения кожи. Диагноз ставят на основании клинической картины и данных лабораторных исследований сыворотки крови и ликвора. Использование диеты с низким содержанием фитиновой кислоты позволяет добиться некоторого восстановления неврологических и зрительных функций.
Асфиксическая дисплазия грудной клетка Жене — аутосомно-рецессивное заболевание, проявляющееся абиотрофией сетчатки (рис. 8.13) и множественными аномалиями скелета, обусловливающими угрожающие жизни дыхательные расстройства у новорожденных и детей раннего возраста.
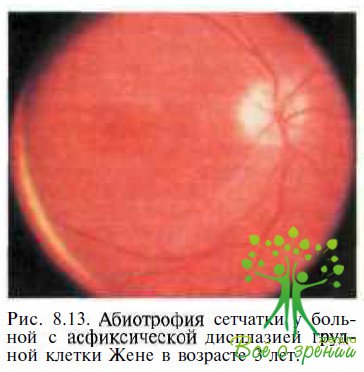
ЭРГ у пациентов с асфиксической дисплазией значительно снижена или отсутствует. Нарушения скелета при синдроме Жене включают карликовость, аномально короткие конечности, полидактилию, лицевой дисморфоз, воронкообразную грудную клетку. У большинства детей нарушения дыхания или присоединение инфекционных осложнений отмечают R возрасте до 1 года. Дифференциальная диагностика с врожденным амаврозом Лебера обычно не представляет трудностей вследствие грубых изменений скелета у пациентов с синдромом Жене.
Оливомостомозжечковая атрофия (ІІ тип по классификации Harding А.Е., 1984, или ІІІ тип по классификации Konigsmark В.М., 1970) — аутосомно-доминантное заболевание, характеризующееся сочетанием пигментной дегенерации сетчатки и признаков мозжечковой атаксии.
Необходимость в проведении дифференциальной диагностики врожденного амавроза Лебера и ОММА III типа возникает лишь в случае позднего выявления (в возрасте 1—3 лет) зрительных расстройств и изменений у ребенка. Офтальмологические поражения включают нистагм, офтальмоплегию, пигментные изменения сетчатки в Макуле и на средней периферии, атрофию зрительного нерва. Ретинальные изменении могут развиваться уже в раннем детском возрасте, а изредка в течение некоторого времени являются единственными симптомами заболевания. В отличие от пациентов с врожденным амаврозом Лебера у больных с ОММА на ранних стадиях болезни ЭРГ может быть нормальной, позднее отмечают снижение амплитуды b-волны и далее — постепенную редукцию обоих компонентов общей ЭРГ и исчезновение ритмической (30 Гц) ЭРГ. Системные проявления ОММА III типа включают атаксию, дизартрию и экстрапирамидную симптоматику.
Диагноз ставят на основании данных клинических и электрофизиологических исследований, а также результатов при которой выявляют атрофию моста и мозжечка.
Лечение. В настоящее время патогенетически обоснованных методов лечения амавроза Лебера существует.
Экспериментируя на мышах с мутацией гена Rpe65, J.P. Hooser и соавт. (2002) обнаружили, что назначение 9-ци-сретинола позволяет длительно сохранять у них зрительные функции. Возможно, аналогичная терапия может быть эффективна у пациентов с мутацией гена RPE65.
Важными аспектами лечения детей с амаврозом Лебера являются их ранняя психологическая реабилитация и педагогическая коррекция связанных со зрительной отклонений в психическом развитии.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0