Х-сцепленный врожденный (ювенильный) ретиношизис


Описание
Х-сцеплснный врожденный (ювенильный) ретиношизис — сцепленная с полом двусторонняя врожденная витреоретинальная дегенерация. Подавляющей большинство таких больных — лица мужского пола. Частота заболевания в популяции — 1 : 10 ООО.Заболевание впервые описано J. Haas и К. Stabsarzt (1898), которые наблюдали «радиальную кистозную макулопатию» у двух мужчин и высказали предположение о том, что заболевание имеет воспалительную природу. Через 15 лет Н.Е. Pagenstecher опубликовал сообщение о семье, у нескольких членов которой был выявлен ретиношизис. В его публикации был показан сцепленный с полом тип наследования заболевания. Несколько позже J. Anderson (1932) и Е. Thomson (1938) описали изменения на периферии сетчатки у пациентов с ретиношизисом.
А. Sorsby и соавт. (1951) дали описание 8 мужчин с ретиношизисом из 3 поколений одной семьи, включающей более 80 родственников, сделав вывод о сцепленным с полом рецессивном характере наследования заболенания. Позанес поя пились многочисленные сообщения о семьях, в которых и 4—7 поколениях были обнаружены больные с Х-сцепленным врожденным ретиношизисом.
Генетика и патогенез. Заболевание передается через Х-хромосому, болеют преимущественно лица мужского пола. Женщины обычно клинически здоровы и являются носителями патологической Х-хромосомы. Их сыновья имеют 50 % шанс унаследовать заболевание, а дочери — 50 % вероятность стать носителями. Н. Forsius и соавт. (1969) обнаружили случай гомозиготного поражения женщины, у отца которой был Х-сцеплснный врожденный ретиношизис, а мать, являющаяся его двоюродной сестрой, оказалась носителем патологического гена. R. Mendoza-Londono и соавт. (1999) описали большую колумбийскую семью, у членов которой наблюдался Х-сцепленный врожденный ретиношизис. Характерные клинические и электрофизиологические изменения выявлены у 27 мужчин и 3 женщин. Тяжесть функциональных и клинических проявлений заболеваний у мужчин и женщин была одинаковой.
Обнаружены другие варианты наследования врожденного ретиношизиса, в частности аутосомно-рецессивный и аутосомно-доминантный. Эти заболевания развиваются у лиц не только мужского, но и женского пола.
Ген RS1, ответственный за развитие ретиношизиса, был картирован в дистальной части короткого плеча Х-хромосомы в интервале Хр22.1 — р22.2. Сцепление гена RS! с двумя полиморфными маркерными генами — DXS43 и DZS41 в Хр22.1 — р22.2 было установлено в исследованиях A. Gal (1988) и N. Dahl и соавт. (1988). G. Gellert и соавт. (1988) при проведении молекулярных генетических исследований у членов 6 семей с Х-сцепленным врожденным ретиношизисом обнаружили, что ген RS1 более тесно сцеплен с маркерным геном DXS9. Авторы предположили следующий порядок расположения гена и маркеров в Хр21.1 — р22.2: RS1 — DXS9 - DXS43 - DXS16 - DXS41.
Ген RS1 образован шестью экзонами, кодирующими 224 аминокислоты белка-предшественника, который включает лидерный пептид, состоящий из 23 аминокислотных остатков, и вы со ко консервативный дискообразные исследования других белков с дискообразными мотивами показали, что этот домен играет важную роль в фосфолипидном связывании и клеточной адгезии. Подобные функциональные свойства могут быть присущи и белку RS1, что коррелирует с патоморфологическими изменениями при ретиношизисе. Мутации, вызывающие изменения структуры белка или его преждевременную деградацию в момент процессинга, приводят к расщеплению сетчатки между слоем нервных волокон и внутренней пограничной мембраной. Другой морфологический дефект — дегенеративные изменения клеток Мюллера, обусловленные нарушением транспорта ионов К+.
Установлено около 100 различных мутаций, приводящих к нарушениям функциональной активности белка RSI и ответственных за развитие Х-сцепленного врожденного ретиношизиса. Они включают внутригенные микроделе- ЦИИ (7 % случаев), нонсенс-мутации (6 %), миссенс-мутации (75 %), мутации в сайте сплайсинга (6 %), а также небольшие вставки и делеции, приводящие к сдвигу рамки считывания (6 %).
Экзоны 1—3 содержат небольшое количество мутаций, приводящих, как правило, к образованию усеченных транслянтов. Это свидетельствует о незначительной функциональной роли этого сегмента в белке. Экзоны 4—6, кодирующие дисковидный домен, содержат наибольшее число мутаций, преимущественно миссенс-мутаций. При их анализе выявлено преобладание мутации Argl97Cys в сайте, имеющем фатальное значение для образования третичной структуры и, следовательно, для функционирования белка. Часто наблюдаются также мутаци и не которых аминокислот, которые, возможно, участвуют в специфичных белок-белковых взаимодействиях на поверхности мембран.
R. Mendoza-Londono и соавт. (1999) выявили делецию одного основания (G) в позиции 639 (639 del G), вызывающую сдвиг рамки считывания при трансляции. В результате этого синтезируется белок с резко увеличенным дисковидным доменом, содержащий 235 аминокислот вместо 224 и превосходящий нормальный по длине.
Несмотря на гетерогенность мутаций, вызывающих ретиношизис, патоморфологические механизмы заболевания у пациентов из разных семей в значительной степени схожи, хотя отмечаются выраженные различия в клинической картине и характере течения. Не выявлено фенотипа врожденного ретиношизиса, который был бы характерен для больных женского пола.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Рефракция, у детей с Х-сцепленным врожденным ретиношизисом обычно бывает гиперметро пня, часто сочетающаяся с астигматизмом. Гиперметро- пия у больных с Х-сцепленным врожденным ретиношизисом вызвана укорочением аксиальной длины глазного яблока.
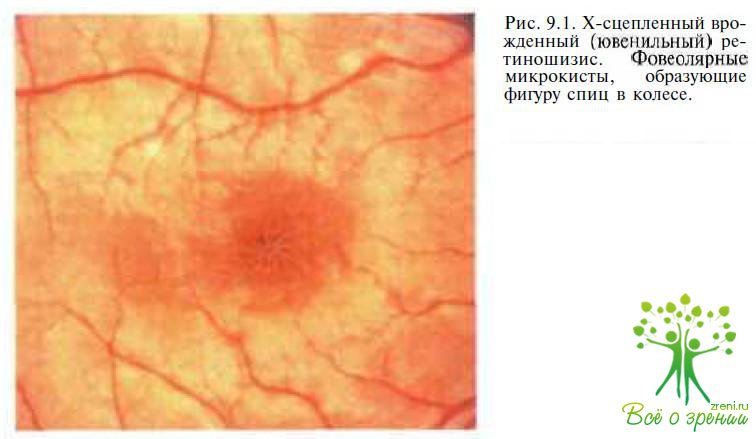
Системные проявления. Фовеолярные изменения, обусловленные наличием микрокист, диагностируют у 98—100 % больных уже в раннем возрасте. При офтальмоскопии в макуле выявляются радиальные складки, называемые симптомом «спиц в колесе» (рис. 9.1).
Позднее в центре формируется углубление (рис. 9.2)
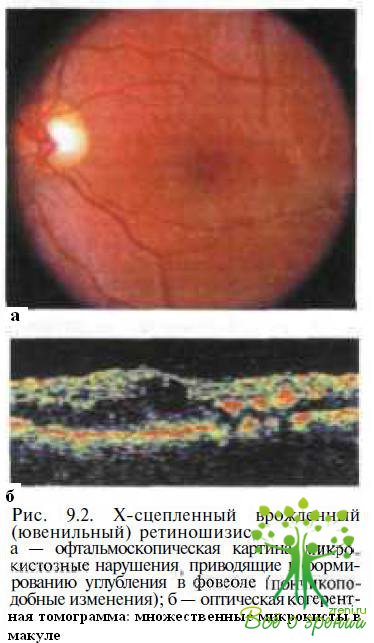
с приподнятыми краями (фигура, напоминающая пончик), а в исходе заболевания — участки хориоретинальной атрофии с отложениями пигмента и твердого экссудата (рис. 9.3).

Иногда в заднем полюсе фиксируют точечные белые очажки, напоминающие изменения при абиотрофии fundus albipunctatus.
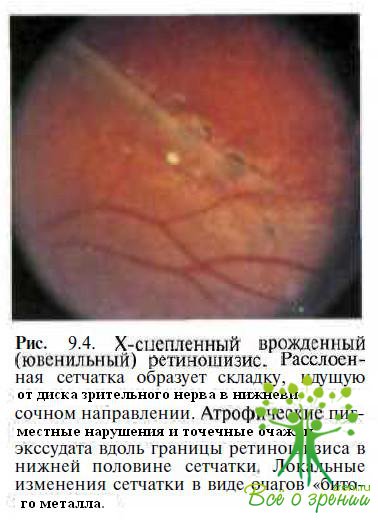
Периферические нарушения, представляющие обширные участки расщепления сетчатки на уровне слоя нервных волокон и локализующиеся преимущественно в нижненаружном квадранте, выявляют у 60—70 % пациентов (рис. 9.4).
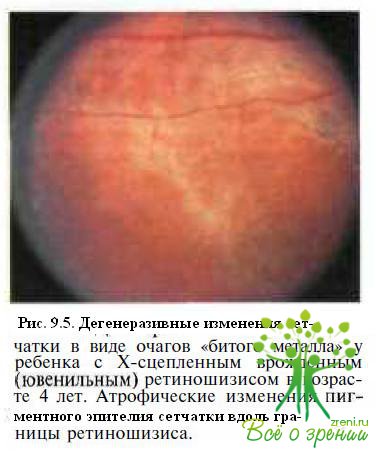
Часто обнаруживают дегенеративные изменения сетчатки в виде рассеянных очажков гиперпигментации и золотисто-серебристых участков, напоминающих поверхность битого металла (рис. 9.5),
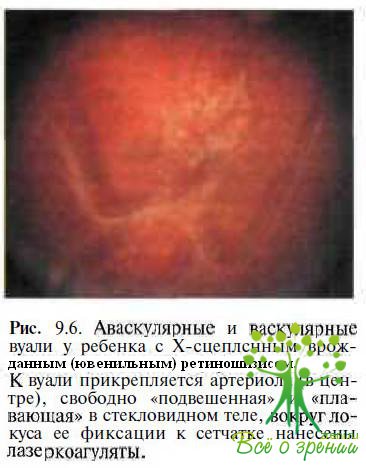
а также аномальные сосуды с повышенной проницаемостью, образующие белые древовидные структуры. В стекловидном теле определяются тяжи, аваскулярные или васкулярные мембраны (рис. 9.6) и вакуоли.
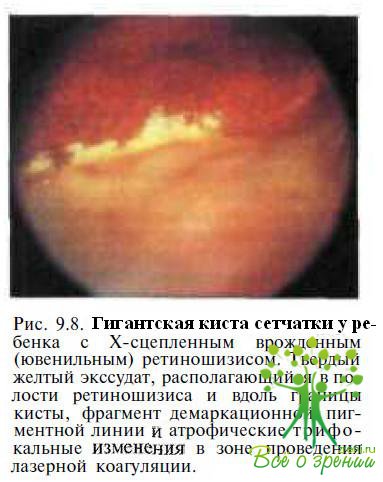
Часто формируются гигантские кисты сетчатки (рис. 9.7),

вдоль границ наблюдается приблизительно у 40 % детей раннего возраста с Х-сцепленным врожденным ретиношизисом и сочетается с косоглазием и нистагмом.
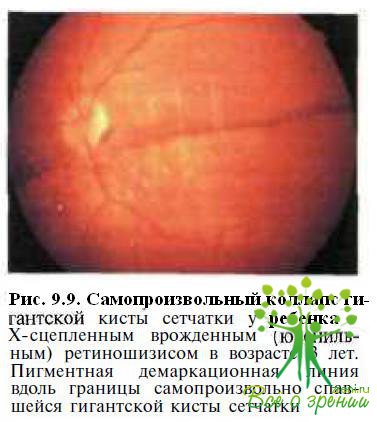
Приблизительно у 85 % детей с буллезной формой Х-сцепленного врожденного ретиношизиса кисты сетчатки самопроизвольно спадают (полностью или частично) через несколько месяцев или лет после момента их возникновения. На месте их первоначальной границы остаются демаркационные пигментные линии (рис. 9.9).
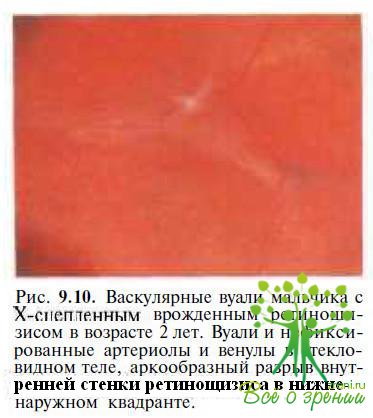
При биомикроскопии можно обнаружить, что ретинальные сосуды нередко как бы подвешены в стекловидном теле (рис. 9.10)
или полостях между слоями расщепленной сетчатки, свободно проходя от слоя нервных волокон к наружным слоям сетчатки. В связи с этим витреоретинальные тракции часто приводят к механическим повреждениям сосудов, вызывая массивные кровоизлияния в стекловидное тело (13—40 % случаев), ретровитреальное пространство или в полости шизиса (20—29 %).
N.D.L. George и соавт. (1995) описали 5 детей в возрасте до 2 лет с нистагмом и/или косоглазием и высоко проминирующим буллезным ретиношизисом, вовлекающим макулу Геморрагии в полость шизиса или стекловидное тело выявлены у 4 больных. При сроке наблюдения от 5 до 6,5 лет во всех случаях отмечено полное прилегание буллезного ретиношизиса с формированием широкой пигментной демаркационной линии, При этом происходит незначительное повышение остроты зрения и уменьшение или исчезновение нистагма.
Автор длительно (от 3 до 11 лет) наблюдал 10 мальчиков с Х-сцепленным врожденным ретиношизисом, косоглазием и нистагмом, у которых в возрасте от 6 до 18 мес были выявлены макулярные изменения и одно- или двусторонние гигантские кисты сетчатки, За период наблюдения в 25 % глаз разнились кровоизлияния и гюретиношизиса или стекловидное тело, которые полностью резорбировались в течение 3—8 мес. В 45 % глаз отмечен самопроизвольный коллапс гигантских кист с формированием в области их первоначальной границы широких демаркационных пигментных линий, В 35 % глаз регрессия кист произошла после лазерной коагуляции сетчатки. При осмотре детей в возрасте 6—12 лет установлено, что острота их зрения составляла 0,05—0,9 (в среднем 0,36 ± 0,23), Нистагм исчез у 4 больных, Ни у одного из 10 детей с буллезным ретиношизясом не было выявлено отслойки сетчатки к моменту завершения наблюдения.
При прогрессировании ретиношизиса в его внутренних слоях появляются множественные аркообразные разрывы (рис. 9.11), развиваются глиальная пролиферация и неоваскуляризация сетчатки, гемофтальм или кровоизлияния в полость кист, витреоретинальные тракции приводят к формированию сквозных разрывов сетчатки.
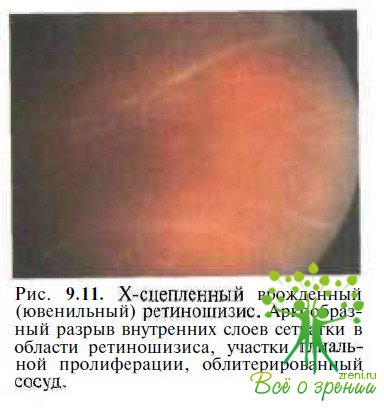
Траклионная или регматогенная отслойка сетчатки развивается у 10— 16 % детей с Х-сцепленным врожденным ретиношизисом. Описаны казуистические случаи развития экссудативной отслойки сетчатки.
Зрительные функции. Острота зрения у детей с Х-сцепленным врожденным ретиношизис ом варьирует от светопроекции до 0,9. Н. Рогаия и соавт. (1973) при обследовании 183 больных с Х-сцепленпым врожденным ретиношизисом определили, что средняя острота зрения у взрослых пациентов составляет 0,34. A.F. Deutman (1977) сообщил о 85-летнем пациенте с Х-сцепленным врожденным ретиношизисом, острота зрения которого составляла 0,03 и 0,05.
М.Т. Roesch и соавт. (1998) длительное время (в среднем 19,78 ± 8,74 лет) наблюдали за естественным течением заболевания у 92 пациентов с ретиношизисом и отмечали постепенное снижение средней остроты зрения с 0,3 при первом исследовании до 0,26 при заключительном осмотре. В 7 % случаев острота зрения у пациентов из этой группы уменьшилась до правильной светопроекции и ниже.
При периметрии выявляются полиморфные дефекты, соответствующие зонам ретиношизиса. Это могут быть квацрантопсия или концентрическое сужение. При статической периметрии порог яркостной чувствительности у большинства пациентов значительно повышен (на 0,6—1,0 log U), обнаруживаются относительные и абсолютные центральные, парацентральные и периферические дефекты.
Диагностика. Диагноз устанавливают на основании данных офтальмоскопии, анализа семейного анамнеза и Вспомогательные методы диагностики — ЭРГ, оптическая когерентная томография, ФАГ И эхография.
При регистрации скотопической и фотопической ЭРГ отмечается выраженное снижение амплитуды Ь-волны, тогда как амплитуда а-волны сохраняется нормальной (рис. 9.12).

Снижение амплитуды b-волны ЭРГ свидетельствует о поражении внутренних слоев сетчатки, в частности клеток Мюллера) что коррелирует с данными морфологических исследований. Преимущественное поражение клеток внутреннего ядерного слоя (биполярных и/или клеток Мюллера) также проявляется значительным снижением амплитуды волны N95 патгерн-ЭРГ. При прогрессировании ретиношизиса в патологический процесс вовлекаются фоторецепторы, что приводит к снижению амплитуды а-волны ЭРГ.
У большинства пациентов с Х-сцепленным врожденным ретиношизисом выявляют значительное снижение амплитуды ритмической выявляют локальной ЭРГ, обусловленное кистозными изменениями в макуле.
ЭОГ в начальной стадии заболевания, как правило, нормальная. Субнормальная ЭОГ встречается у пациентов в терминальной стадии болезни, сочетаясь со значительно редуцированной ЭРГ.
При диагностике заболевания используют электрофизиологические методы, обладающие высокой чувствительностью. Нарушения ЭРГ в виде снижения амплитуды и/или задержки латентности b-волны фиксируют у 90—95 % носителей, не имеющих изменений на глазном дне.
При флюоресцентной ангиографии в начальной стадии процесса изменений в макуле нет. По мере прогрессирования заболевания отмечают центральную шпер- флюоресценцию из-за дефектов пигментного эпителия сетчатки, а также повышенную проницаемость флюоресцсина через стенки аномальных и новообразованных сосудов.
Дифференциальная диагностика.
В зависимости от тяжести витреоретинальных изменений необходимо дифференцировать Х-сцепленный врожденный ретиношизис от ретинопатии недоношенных, болезни Гольдманна— Фавра, болезни Вагнера, синдрома Стиплера, болезни Норри, семейной экссудативной витреоретинопатии, болезни Штаргардта, пигментного ретинита.
Ретинопатия недоношенных. Диагностика может вызывать затруднения при осмотре детей с регрессивной ретинопатией недоношенных III— IV стадий. У таких детей определяют диффузные пигментные нарушения, атрофические хориоретинальные очаги, области фиброзной пролиферации (рис. 9.13)
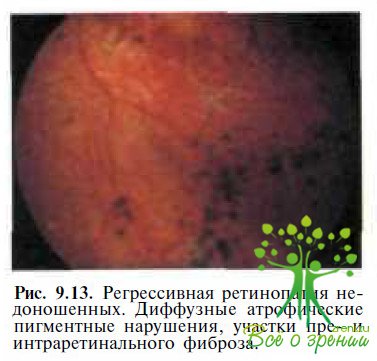
и тракционную отслойку сетчатки. Диск зрительного нерва обычно деформирован, нейроретинальный край диска, сосудистый пучок и макула эктопированы в нижненаружном направлении (рис. 9.14),
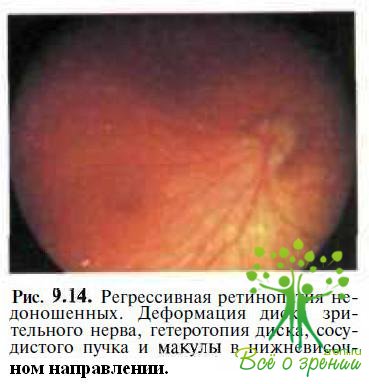
где на периферии выявляются зоны фиброза. ЭРГ у пациентов с ретинопатией недоношенных существенно снижена, иногда не регистрируется. Существенное значение для дифференциальной диагностики имеют результаты нейросоматического обследования и данные анамнеза (гестационный возраст детей при рождении меньше 29 нед, масса тела — меньше 1500 г).
Болезнь Гольдманна-Фавра передается по аутосомно-рецессивному типу, поражает как мужчин, так и женщин. Стекловидное тело при биомикроскопии выглядит оптически пустым, в нем нет вуалей, но формируются лентоподобные тяжи. На периферии сетчатки определяются выраженный ретиношизис и пигментные нарушения, в макуле — грубые кистозные изменения. Рано развивается катаракта. ЭРГ отсутствует. В начале заболевания характерны жалобы на снижение сумеречного зрения, позднее развивается тяжелая никталопия. При периметрии выявляется концентрическое сужение границ поля зрения.
Болезнь Вагнера — редко встречающаяся витреоретинальная дистрофия с аутосомно-доминантным типом наследования, характеризующаяся постепенным снижением остроты зрения и прогрессирующим сужением поля зрения. Изменений в макуле обычно не бывает, поэтому острота зрения у пациентов достаточно высокая. В стекловидном теле выявляют белые васкуляризированные мембраны, локализующиеся преимущественно в экваториальной зоне. На периферии определяется ретиношизис. Отслойка сетчатки развивается крайне редко. Уже к 10-летнему возрасту появляются помутнения кортикальных слоев хрусталика, которые быстро прогрессируют. ЭРГ, как правило, субнормальная.
Семейная экссудативная витреоретинопатия. Преодолеть возникающие в некоторых случаях диагностические трудности, особенно у больных с X- сцепленным типом наследования экссудативной витреоретинопатии, помогают результаты офтальмоскопии (наличие серповидной складки сетчатки и гетеротопии макулы и диска зрительного нерва, отсутствие у большинства пациентов кистозной макулопатии) и ФАГ (выявление зон и сосудистых нарушений в виде «морского веера» на границе аваскулярной и васкулярной областей сетчатки у больных с семейной экссудативной витреоретинопатией). При аутосомно-доминантном типе наследования экссудативной витреоретинопатии важное диагностическое значение имеют данные семейного анамнеза и/или анализа ДНК.
Болезнь Штаргардта передается но аутосомно-рецессивному типу. Поражаются одинаково часто как мальчики, так и девочки. В макулярной области выявляют минимальные дистрофические изменения (рис. 9.15).
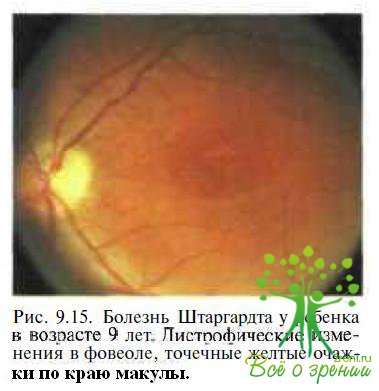
В отличие от детей с Х-сцепленным врожденным ретиношизисом, у которых острота зрения снижена уже с рождения, у пациентов с болезнью Штаргардта в возрасте до 6—8 лет острота зрения может быть высокой. По мере прогрессирования процесса она снижается. На ранних стадиях заболевания только регистрация ЭРГ позволяет дифференцировать болезнь Штаргардта от Х-сцепленного врожденного ретиношизиса. У детей с болезнью Штаргардта амплитуда b-волны ганцфельд ЭРГ нормальная, у больных с Х-сцепленным врожденным ретиношизисом — значительно снижена. При периметрии у детей с болезнью Штаргардта определяется центральная скотома.
Болезнь Илза. Дифференциальная диагностика может вызывать определенные трудности в развитой стадии заболевания, для которой характерны рецидивирующие гемофтальмы, помутнения стекловидного тела в виде мембран, неоваскуляризация сетчатки и витреоретинальная пролиферация. Болезнь Илза чаще развивается у мужчин молодого возраста. У 17—48 % пациентов отмечают вестибулярные и слуховые дисфункции. Болезнь Илза распространена на территории Ближнего Востока и крайне редко встречается в России. У пациентов с болезнью Илза в отличие от детей с Х-сцепленным врожденным ретиношюисом первые симптомы появляются в возрасте 12—30 лет, макуладлительно сохраняется интактной, характерна высокая острота зрения — 60 % больных имеют конечную остроту зрения 0,5 и выше.
Серповидная (фальциформная) отслойка сетчатки — заболевание, описанное в литературе также под названием «фальциформпые складки сетчатки», — легкая форма ретинальной дисплазии, наследуемая по аутосомно-рецессивному типу. Обычно складки сетчатки простираются от диска зрительного нерва к парному телу в нижненаружном квадранте. Вдоль складок отмечаются пигментные изменения, у большинства больных — персистирующие гиалоидные сосуды. Мембран и вуалей в стекловидном теле у пациентов с серповидной отслойкой сетчатки не бывает.
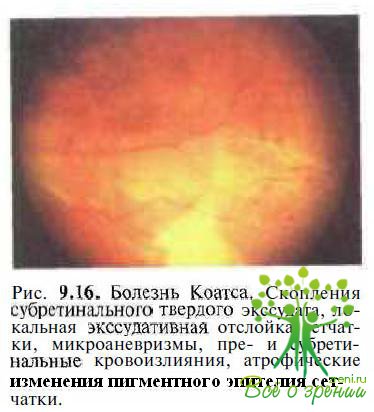
Болезнь Коатса. У детей раннего возраста с буллезной формой Х-сцеп- ленного врожденного ретиношизиса нередко определяют накопление твердого экссудата в полости шизис и в субретинальном пространстве, периферические полиморфные сосудистые изменения в виде периваскулярных муфт, неоваскуляризации, микроаневризм и облитерированных сосудов. В ряде случаев заболевание осложняется гомофтальмом, формированием преретинальных фиброглиальных масс, развитием экссудативной отслойки сетчатки. Описаны казуистические случаи развития неоваскулярной глаукомы у детей раннего возраста с Х-сцепленным врожденным ретиношизисом и тотальной отслойкой сетчатки.
Решить возникающую в таких случаях диагностическую дилемму помогают офтальмоскопические симптомы (радиальная исчерченность в макуле, вуали в стекловидном теле) и данные анамнеза, указывающие на сцепленный с полом характер наследования заболевания. При регистрации скотопической и фотопической ЭРГ у пациентов с Х-сцспленным врожденным ретиношизисом выявляется селективное снижение амплитуды Ь-волны.
У 98 % детей с болезнью Коатса поражается один глаз. Заболевание спорадическое. При офтальмоскопии в макуле выявляют желтый Лроминирующий очаг в связи с субретинальным накоплением твердого экссудата, на периферии — сосудистые мальформации (рис. 9.16). У пациентов с болезнью Коатса в начальной стадии ЭРГ не изменяется, в развитой и далекозашедшей стадиях ЭРГ пораженного глаза субнормальная или не регистрируется, интактного глаза — нормальная.
Пигментный ретинит. Диагностические трудности могут возникать только при обследовании детей с Х-сцепленным врожденным ретиношизисом, у которых нет изменений стекловидного тела и периферических отделов сетчатки. Диагноз устанавливают на основании результатов офтальмоскопии, регистрации ЭРГ, оптической когерентной томографии, анализа анамнестических данных и молекулярных генетических исследований.
Х-сцепленная форма пигментного ретинита характеризуется диффузными или локальными пигментными нарушениями сетчатки, побледнением диска зрительного нерва, сужением ретинальных сосудов.(рис. 9.17).
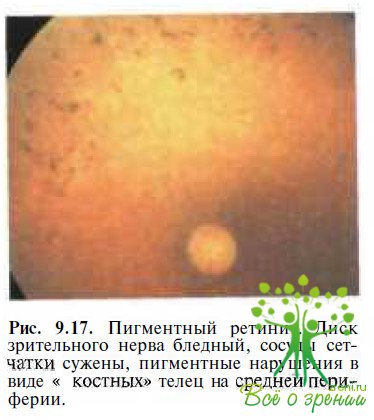
Иногда возникает кистозный макулярный отек, Острота зрения начинает снижаться в возрасте 6—8 лет или старше, ухудшается ночное зрение, при периметрии выявляют концентрическое сужение периферических границ поля зрения или верхневисочную квадрантопсию, корреспондирующую с пигментными нарушениями в нижненосовом отделе сетчатки, Электроретинограмма значительно редуцирована или отсутствует. Известны два основных генетических локуса, ответственных за развитие заболевания. Ген RP2 сцеплен с LI. 28 винтер вале Хр 11.3, ген RP3 идентифицирован в позиции Хр21.
Лечение. В начальной стадии заболевания проводят коррекцию аметропии, лечение амблиопии и косоглазия, рекомендуется использование препаратов, улучшающих микроциркуляцию и обменные процессы в сетчатке. Критериями регрессии врожденного ретиношизиса являются спонтанный коллапс ретинальных кист, ретракция витреальных мембран и отрыв их от места прикрепления к сетчатке, облитерация новообразованных сосудов, резорбция экссудата и геморрагий, стабилизация остроты зрения.
При прогрессируюшем течении болезни применяют лазерную коагуляцию сетчатки. Признаками прогрессирования процесса являются снижение остроты зрения, распространение ретиношизиса по площади или высоте, неоваскуляризация сетчатки, появление разрывов во внутренних и/или наружных слоях ретиношизиса или увеличение их размеров, кровоизлияния в стекловидное тело или полость шизиса, развитие отслойки сетчатки.
Лазерную коагуляцию проводят в несколько этапов. Первоначально наносят коагуляты вдоль границ ретиношизиса, вокруг мест прикрепления шварт к сетчатке и зон, где имеются аномальные или новообразованные сосуды. Затем коагулируют зоны неоваскуляризации и, по возможности, поверхность ретинальных кист. Диаметр коагулятов при барьерной профилактической лазерной коагуляции составляет 200—300 мкм, экспозиция — 0,05—0,1 с, мощность — 90— 200 мкВ.
R.J, Brockhurst (1970) считает нецелесообразным проведение лазерной коагуляции сетчатки больным с X- сцепленным врожденным ретиношизисом без признаков неоваскуляризации.
В случае развития отслойки сетчатки, распространении кисты на макулу и выраженной экссудации используют экстрасклеральное пломбирование, витрэктомию, интравитреальную тампонаду перфторуглеродами или силиконовым маслом.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0