Нейрофиброматоз


Содержание:
Описание
В настоящее время принято считать, что нейрофибромагоз включает как минимум две клинически различающиеся аутосомно-доминантные формы: нейрофиброматоз 1-го типа (болезнь Реклингхаузена) и нейрофиброматоз 2-го типа (двусторонняя невринома слухового нерва). Некоторые авторы выделяют также сегментарную форму. Одним из вариантов семейной формы нейрофиброматоза 1-го типа является синдром Уотсона (Watson). У детей с этой формой заболевания выявляются легочный стеноз, небольшой рост, кожные изменения, меланоцитарные гамартомы радужки, отставание в психическом развитии и относительная макроцефалия. У представителей нескольких родословных с синдромом Уотсона была выявлена делеция гена NFJ .↑ Нейрофиброматоз 1-го типа (болезнь Реклингхаузена)
Нейрофиброматоз 1-го типа (-1) — мультисистемная эктодермальная дисплазия с аутосомно-доминантным типом наследования и высоким уровнем мутаций.
Эпидемиология и генетические исследования. Нейрофиброматоз встречается в популяции с частотой 1 : 3000 — 4000 у новорожденных. Заболевание наследуется по аутосомно-доминантному типу. Доля спорадических случаев составляет 35 — 50 %. Ген, ответственный за развитие нейрофиброматоза-1, картирован в перицентрической области проксимальной части короткого плеча 17-й хромосомы в локусе 17ql 1.2. В 1991 г. была полностью расшифрована последовательностъ гена нейрофиброматоза-1 (NF1), кодирующего продукт, названный «нейрофибромин», охарактеризованы его клеточная экспрессия и функция.
Ген нейрофиброматоза-1 перекрывает область геномной ДНК в 350 kb (kb— килобаза, равная 1 тыс. пар нуклеотидов) и является одним из самых крупных генов, кодирующих заболевания человека. Ген содержит 59 экзо- нов, которые после транскрипции формируют мессенджер РНК в 13 kb. В образовании нейрофибромина участвуют 8454 нуклеотвда. Собранный из 2818 аминокислот нейрофибромин, по-видимому, является цитоплазматическим белком. Хотя ген нейрофиброматоза-1 экс пресс ируется повсеместно, он может иметь специализированную функцию в клетках нервного гребня. Предполагают, что он связан с формированием цитоскелета.
В некоторой степени курьезно, что три малых гена ( OMgp, EVI2A, EV12B), которые читаются в противоположном направлении от конца большого гена нейрофиброматоза-1, расположены внутри одного его интрона (некодирующей области). Этот небольшой «ген в гене» достаточно интересен, поскольку известны значение OMgp (от «олигодендроцит, миелин, гликопротеин») в межклеточных коммуникациях в центральной нервной системе и роль EV12A, EVI2В в развитии мышиной лейкемии. Возможно, они являются «пережитком прошлого», т.е. остатками наследства от филогенетически более древних генов. Однако нет доказательств, что мутации этих внедренных генов могут вызывать какие-либо специфические фенотипы нейрофиброматоза-1.
Область GAP в гене нейрофиброматоза-1, Полагают, что главной активной порцией белка нейрофиброматоза-1 является область GAP [гуанозинтрифосфат (ГТФ)-активирующий белок], названная так за ее поразительную последовательность, гомологичную каталитическому домену GAP млекопитающих, и эквивалентные дрожжевые белки IRAI и IRA2. GAP кодирует GAP-подобные белки, которые могут действовать как регуляторы роста, взаимодействующие с онкогеном ras (вирусом саркомы крыс). Сам по себе GAP — важный для клеточного цикла регуляторный белок, взаимодействующий с клеточным онкогенным ras и катализирующий конверсию активной ГТФ-связанной формы ras в неактивную. Однако, если ген ras мутирует, белок ras теряет способность связываться с ГТФ и может продолжать активировать клетку, утрачивая важный механизм контроля. Действительно ли ras контролирует GAP или все происходит наоборот, пока неизвестно.
Эмбриональные мутации в гене NF1. Следующим шагом после клонирования гена заболевания является определение типов и консистенции мутаций, ответственных за развитие болезни, и существования фенотипически-генотипических корреляций. В настоящее время идентифицировано несколько типов зародышевых линий мутаций у больных с нейрофиброматозом-1: мегаделеции, которые могут приводить к развитию нейрофиброматоза, сочетающегося с другими нарушениями (например, с олигофренией или фенотипом Ноунана), микроделеции, точечные мутации, вставки или транс локации. Хотя и были обнаружены случайные совпадающие мутации в гене нейрофиброматоза-1 у больных из разных семей, подобное явление нельзя считать особенностью данного заболевания. Отсутствие «горячих» мутационных точек (отрезки хромосом с более высокой концентрацией мутаций, чем на остальных участках) является особенностью опухолевых сулрессорных генов. Интересно, что большинство новых зародышевых линий мутаций возникает на отцовской хромосоме, являясь, возможно, отражением дефектов в периоде сперматогенеза. Зависимость частоты мутаций от такого фактора, как возраст отца, до конца не изучена.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Механизм формирования опухоли при нейрофиброматозе-1. Рост и дифференцирование клеток организма контролируется двумя типами генов, подобно тому как управление автомобилем осуществляется с помощью педалей акселератора и тормоза. Гены, обеспечивающие рост и дифференцировку, называют протонкогенами или онкогенами, а комплементарные им гены, ингибирующие эти процессы, — опухолевыми (илиростовыми) супрессорнымигенами. Ряд клеточных онкогенов известен. К ним относятся гены ras, myc, sre, fos и erb обладающие способностью постоянно активироваться и обеспечивать неконтролируемую клеточную пролиферацию. Следовательно, можно представить, в слегка упрощенном виде, что возникновение опухолей опосредуется двумя сочетающимися процессами: инактивирующими мутациями опухолевых сулрессорных генов (например, в случае ретинобластомы) или активирующими мутациями клеточных онкогенов. Наилучшим доказательством того, что ген NF1 действует как опухолевый супрессорный ген, было бы обнаружение второй мутации или делеции в другой «нормальной» копии гена NF1 в клетках опухолей, а также наследственной мутации в гене NF1. Если же он действует как клеточный онкоген, то никакой мутации не может быть обнаружено. При первом варианте развитие опухолей было бы рецессивным явлением, при втором — доминантным.
Парадигм ретинобластомы является подходящей моделью механизма онкогенеза в его наиболее простой форме. Для развития опухоли требуется мутация в гене ретинобластомы (Rb), следующая за соматической мутацией в ретинальной клетке в локусе ретинобластомы, или две отдельные соматические мутации в двух аллелях гена Rb в такой же клетке. При сравнении конституционной ДНК с опухолевой ДНК у одного индивидуума может быть обнаружена потеря аллеля, Это вызвано существованием полиморфизмов ДНК генома всех индивидуумов, которые приводят к различиям в длине выделенных материнских и отцовских копий любой хромосомы после рестриктазного расщепления, разделения гельэлектрофорезом и блоттинга по Саузерну. Потеря одной из структур в опухолевой ДНК может быть идентифицирована при сравнении ее с конституционной ДНК в лейкоцитах. Данное явление называется потерей гетерозиготности. Для более точного определения маленьких генетических мутаций или перестроек необходимо проводить секвенированис ДНК.
Если опухоли, развивающиеся при нейрофиброматозе-1, ведут себя подобно ретииобластоме, то в опухолевой ДНК по сравнению с конституционной ДНК лейкоцитов могла бы быть обнаружена потеря всего или части аллеля 17-й хромосомы, соответствующего гену МВТ. Данная гипотеза послужила поводом для проведения анализа ДНК доброкачественных опухолей у больных с нейрофиброматозом-1, в ходе которого не удалось выявить никаких макроскопических делеций в плексиформных нейрофибромах, глиомах зрительного нерва или нейрофибромах ствола мозга. Между тем при исследовании 22 нейрофибросарком у пациентов с нейрофибромато¬зом-1 обнаружили потерю маркера 17р (короткое плечо 17-й хромосомы, где расположен генр53) или маркеров 17р и 17q (длинное плечо), включая интервал NFT в пяти и шести случаях соответственно. Таким образом, у 50 % пациентов с нейрофибросаркомами была выявлена потеря 17р, содержащего ген р53, что, возможно, является критическим звеном в генезе этих опухолей. Белок p53 — регулятор клеточного цикла, действующий в G- фазе репликации, вероятно, на уровне транскрипции или инициации репликации генов, имеющих значение для регуляции клеточного цикла. Однако некоторые исследователи считают, что обе копии гена NF1 могут быть нарушены в нейрофибросаркомах, феохромоцитомах и злокачественных невриномах. При анализе ДНК в злокачественных астроцитомах также было отмечено отсутствие маркеров 17ри 17q либо только 17q. Двойная делеция гена NFI в определенных локусах является предварительным доказательством того, что он ведет себя как опухолевый супрессорный ген.
Системные проявления. Наиболее часто наблюдаемые проявления нейрофиброматоза-1 — поражения кожи, представляющие собой различные типы нарушения ее пигментными, и нейрофибромы. Пятна на коже цвета «кофе с молоком» — плоские участки гиперпигментации диаметром от I — 2 ММ ДО Уз ем — появляются вскоре после рождения у 99 % пациентов с нейрофиброматозом-1 (рис. 11.1).
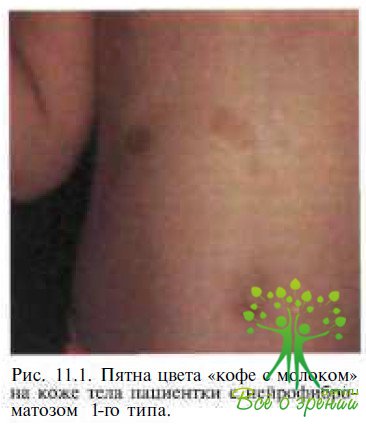
Несколько реже наблюдается гиперпигментация в виде веснушек или невусов, а также пигментные пятна, располагающиеся поверх проминирующих нейрофибром. Количество пигментных изменений кожи при нейрофиб-роматозе-1 значительно варьирует: от 2—3 мелких пятнышек до нескольких сотен участков пигментации различного диаметра. Как правило, с возрастом количество и площадь зон гиперпигментации увеличиваются.
Нейрофиброма — опухоль, связанная с оболочками нерва и представляющая собой комбинацию клеточных элементов, имеющихся в нормальной коже, таких как фибробласты, меланоциты, нервные, шванновы и тучные клетки. Нейрофибромы могут увеличиваться в пубертатном периоде или во время беременности.
Лабораторных тестов для диагностики нейрофиброматоза нет, и диагноз устанавливают на основании имеющихся характерных симптомов. Диагностические критерии нейрофиброматоза-1 можно представить в следующем виде.
Диагностические критерии нейрофиброматоза 1-го типа
Диагноз нейрофиброматоза 1-го типа устанавливают, если у обследуемого выявляют 2 симптома и более из представленных ниже:
- шесть и больше пятен цвета «кофе с молоком на коже»:
- Диаметр каждого из этих пятен должен быть не менее чем 5 в препубертатном возрасте;
- Диаметр нейменшего из пятен должен быть не менее 15 мм в препубертатном возрасте;
- Диаметр каждого из этих пятен должен быть не менее чем 5 в препубертатном возрасте;
- две или более нейрофибромы любого типа или одна плексиформная нейрофиброма;
- веснушки в подмышечной или паховой областях;
- глиома зрительного нерва;
- две или более гамартомы радужки (узлы Лиша);
- характерные костные нарушения: дисплазия малого крыла клиновидной кости; дугообразное искривление, иногда сочетающееся с псевдоартрозом большеберцовой и малоберцовой костей; деформация, истончение и кисты длинных трубчатых костей и др.;
- наличие родственника первой степени родства (родители, сибсы, по¬томки) с нейрофиброматозом-1.
Офтальмологические проявления. Наиболее часто выявляемые офтальмологические признаки нейрофиброматоза- I — плексиформная нейрофиброма век, меланоцитарные гамартомы радужки, глиома зрительного нерва, астроцитарная гамартома сетчатки, утолщение и проминенция роговичных нервов, конъюнктивальная нейрофиброма, пульсирующий экзофтальм, спиралевидные мальформации венул сетчатки и ее ишемические поражения, пятна цвета «кофе с молоком» на глазном дне, буфтальм, развитие которого связано с хороидальной гамартомой или аномалиями трабекулярной сети.
Плексиформная нейрофиброма век (рис. 11.2)
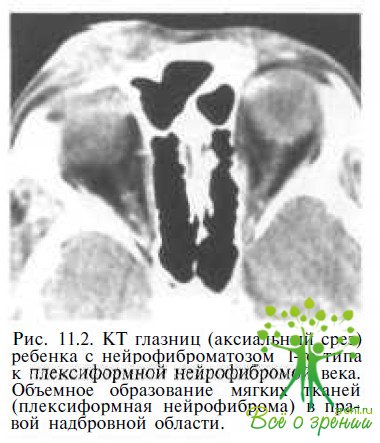
обычно развивается в возрасте от 2 до 5 лет, вызывая S-образный птоз и отек верхнего века. Нейрофиброма века отмечается у 5— 16 % пациентов с нейрофиброматозом- 1. Иногда плексиформная нейрофиброма век сочетается с лицевой асимметрией или ген и гипертрофией. У 50 % пациентов с плексиформной нейрофибромой век развивается ипсилатеральная глаукома.
Меланоцитарные гамартомы радужки (чаще обозначаются эпонимом «узлы Лиша») — один из наиболее специфических симптомов нейрофиброматоза-1. Узлы Лиша представляют собой возвышающиеся над поверхностью радужки образования (рис. 11.3).
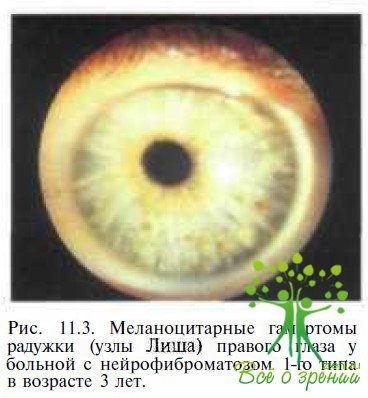
Количество и размеры их варьируют (от одиночных «крупиц соли», с трудом выявляемых при биомикроскопии, до множественных крупных узлов, достигающих 2 мм в диаметре). Цвет гамартом зависит от окраски радужки. У пациентов с голубой или зеленой радужкой узлы Лиша серо-коричневые с пушистыми краями. В тех случаях, когда радужка коричневого цвета, гамартомы кремовые, имеют куполообразную форму и четко определяемые края. Иногда узлы Лиша являются единственным проявлением заболевания. В момент рождения их определяют крайне редко, но уже в возрасте 2,5 года узлы выявляют у 33 % детей с нейрофиброматозом-1, в возрасте 5лет — у 50%,а в 15 лету 75 %. В возрасте от 25 до 35 лет меланоцитарные гамартомы радужки обнаруживают у 96—100 % пациентов с нейро¬фиброматозом-1, причем у 93 % — в обоих глазах.
Глаукома развивается приблизительно y 25 % больных с нейрофибро¬матозом-1 вследствие аномалий трабекулярной сети, механических нарушений взаиморасположения структур угла передней камеры из-за компрессии новообразованиями цилпарного тела или хоровдеи.
Частота развития глиомы зрительного нерва и/или хиазмы у пациентов с нейрофиброматозом-1 точно не установлена и варьирует, по данным литературы, от 2 до 50 %. В детском возрасте глиомы зрительного нерва составляют 2—5 % от общего числа новообразований головного мозга, причем у 33—70 % пациентов с глиомой зрительного нерва выявляют нейрофиброматоз-1. Развитие двусторонних глиом отмечено только у больных с нейрофиброматозом-1. При нейрофиброматозе-1 глиома зрительного нерва чаще развивается в дошкольном возрасте, тогда как идиопатические глиомы зрительного нерва манифестируют обычно в среднем в 12 лет. Первыми функциональными и клиническими симптомами глиомы зрительного нерва являются постепенное снижение остроты зрения, нарушения в поле зрения, отек диска зрительного нерва. По мере прогрессирования заболевания развиваются птоз верхнего века, экзофтальм и атрофия зрительного нерва.
Хиазмальные глиомы прогностически менее благоприятны и образуются у пациентов с нейрофиброматозом-1 чаще, чем глиомы зрительного нерва. В этом случае при офтальмоскопии нередко выявляют атрофию зрительного нерва. В этом случае при офтальмоскопии нередко выявляют атрофию зрительного нерва.
Известны случаи спонтанной полной либо частичной регрессии глиомы зрительного нерва и/или хиазмы у пациентов с нейрофиброматозом-1, подтвержденные результатами МРТ и патогистологических исследований. Механизмы, ответственные за исчезновение опухолей, точно не установлены. Возможными причинами регрессии опухоли могут быть эндокринные нарушения, приводящие к уменьшению наполнения сосудов глиомы или резорбции мукосубстанций, секретируемых глиомой, а также некроз клеток опухоли в ответ на повышение иммунной активности или запрограммированная гибель (апоптоз) клеток.
Частыми осложнениями нейрофиброматоза являются сосудистые нарушения [спиралевидные мальформации венул второго или третьего порядка (рис. 11.4, а),
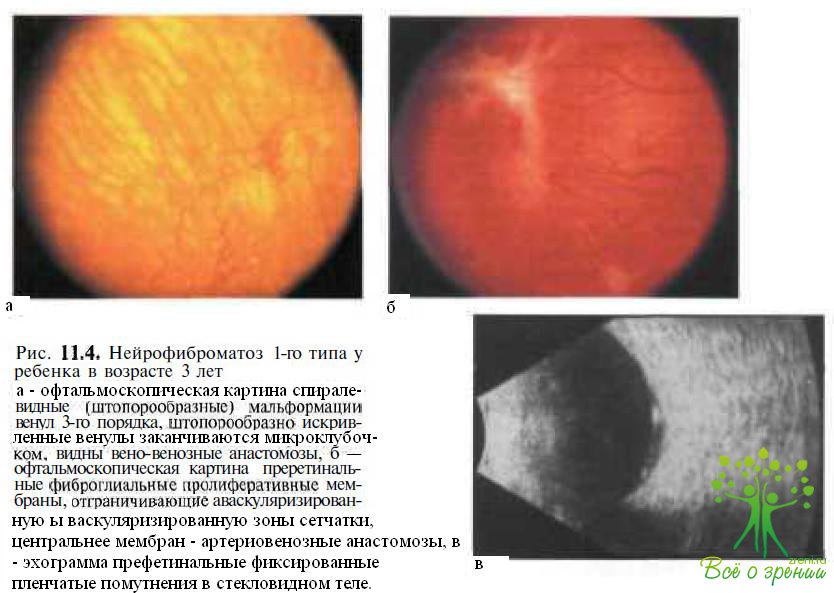
завершающиеся формированием микроклубочков, стеноз мелких сосудов, признаки капиллярной ишемии и формирование компенсаторной коллатеральной циркуляции] в различных органах, в том числе в глазах Предполагают, что на фоне дисплазии гладкомышечных клеток, образующих сосудистую стенку, прогрессирующий рост шванновых клеток приводит к постепенному сужению просвета сосуда, в итоге завершаясь его полной окклюзией. В дальнейшем развиваются периваскулярная пролиферация и фиброз, прогрессирующий рост глиальных клеток. Наиболее характерными проявлениями ишемических нарушений в глазу при нейрофиброматозе-1 являются неперфузируемые области на периферии сетчатки, формирование веновенозных и артериовенозных шунтов и преретинальных фиброглиальных мембран на границе аваскулярных зон (рис. 11.4, б, в), атрофия зрительного нерва.
Спиралевидные (штопорообразные) мальформации ретинальных венул второго или третьего порядка выявляются при офтальмоскопии приблизительно у 38 % больных с нейрофиброматозом-1. Эти микрососудистые аномалии могут проявляться в виде «усеченных» форм, при которых поражается лишь одна венула. Чаще данные аномалии характеризует вовлечение нескольких венул. Обычно они локализуются в височной половине сетчатки на расстоянии 1—2 РД от диска зрительного нерва. Венулы причудливо изгибаются, приобретая штопорообразный ход, а затем постепенно теряются из вида или заканчиваются микроклубочком (см. рис. 11.4, а). Нередко в этих зонах образуются веновенозные анастомозы. Данные изменения лучше визуализируются при флюоресцентной ангиографии.
В нескольких сообщениях, авторы которых использовали сканирующий лазерный офтальмоскоп, описаны хороидальные изменения, выявляемые у 100 % больных с нейрофиброматозом-1: яркие светлые пятки, видимые в инфракрасном свете (780 нм), но отсутствующие при исследовании в режиме гелий-неонового излучения (633 нм). Учитывая высокую частоту хороидальных нарушений, авторы считают целесообразным использовать данный симптом в качестве одного из диагностических критериев нейрофиброматоза 1-го типа.
Лечение. Опухоли, рост которых приводит к деформации окружающих тканей и функциональным нарушениям, подлежат хирургическому лечению. Узлы Лиша не представляют опасности, так как не способствуют развитию глаукомы. Выбор метода лечения глаукомы у больных с нейрофиброматозом-1 зависит от причин ее развития.
Наибольшие противоречия связаны с тактикой лечения глиом хиазмы. Считают, что оптимальными являются консервативное лечение и лучевая терапия. Хирургическое лечение применяют у больных с опухолями больших размеров или опасной локализации, если их прогрессирование доказано с помощью КГ и MPT.
Генетическое консультирование. Поскольку нейрофиброматоз-1 наследуется по аутосомно-домииантному типу с пенетрантностью, близкой к 100 %, то вероятность рождения больного ребенка у пациента с нейрофиброматозом-1 составляет 50 %. Экспрессивность заболевания значительно варьирует: около 25 % больных имеют поражения средней HJIH тяжелой степени, и у остальных определяют лишь отдельные симптомы нейрофиброматоза-1, свидетельствующие о легкой форме болезни. Пренатальная диагностика заболевания с высокой точностью может быть осуществлена лишь в семьях пациентов с нейрофиброматозом-1, у членов которых идентифицированы мутации в локусе 17ql 12 хромосомы.
↑ Нейрофиброматоз 2-го типа
Нейрофиброматоз 2-го типа (-2) — редко наблюдаемое заболевание: частота его развития в популяции приблизительно I : 33 000 - I : 50 000.
Генетические исследования. Ген, ответственный за развитие нейрофиброматоза-2, локализуется на проксимальной части длинного плеча 22-й хромосомы между 22qll.21 и 22ql3.1. Ген-кандидат кодирует белок из 587 аминокислот. Он получил название «merlin» из-за сходства с moesin, ezrin и radixin — тремя членами семейства белков, которые связывают компоненты цитоскелета с клеточными момбриинммн белками. Niep ?IH (называемый также шванномин) функционально представляет собой белок, оказывающий супрессивное воздействие на опухолевый рост. Мутации в таком белке могут влиять на некоторые клеточные процессы, включая деление клетки, межклеточное сообщение, внутриклеточную организацию, сцепление.
Системные проявления и диагностические критерии. Наиболее характерными признаками нейрофиброматоза-2 являются двусторонняя невринома слухового нерва, обычно развивающаяся в возрасте 20—30 лет, а также различные новообразования головного и спинного мозга (менингиомы, шванномы, астроцитомы, нейрофибромы). Диагностические критерии для идентификации нейрофиброматоза-2, установлеииые в 1987 г., можно представить в следующем виде.
Диагностические критерии нейрофиброматтй 2-го типа
Диагноз нейрофиброматоза 2-го типа устанавливают, если у обследуемого выявляют:
- двустороннее новообразование, вовлекающее слуховой нерв (VIII пара черепных нервов), выявляемое с помощью КТ или МРТ; или сочетание признаков:
- наличие родственника в первом колене (родители, сибсы) с нейрофиброматозом-2;
- односторонняя опухоль слухового нерва;
- два симптома из перечисленных ниже:
- менингиома,
- шваннома,
- ювенильные задние субкапсулярные, капсулярные, кортикальные или комбинированные помутнения хрусталика.
- менингиома,
Офтальмологические проявления. Аномалии глаз определяют у 86—87 % пациентов с нейрофиброматозом-2. Частота и тяжесть как соматических, так и глазных нарушений при нейрофиброматозе-2 увеличивается с возрастом. D.G.R. Evans и соавт. (1992) обнаружили клинические проявления нейрофиброматоза-2 у 10 % больных в возрасте 10 лет, у 50 % — 20 лет и у 80 % - 30 лет. Среди офтальмологических проявлений нейрофиб- роматоза-2 можно выделить ювенильные задние субкапсулярные, капсулярные, кортикальные или комбинированные помутнения хрусталика, диагностируемые в 50—85 % случаев, ретинальные гамартомы — 9—22 %, эпиретинальные мембраны — 8—50 глиому или менингиому зрительного нерва — 8—17 %. Несколько реже у пациентов с нейрофиброматозом-2 выявляют хороидальные гамартомы, гипертрофию роговичных нервов, конъюнктивальные нейрофибромы.
Хотя и считают, что меланоцитарные гамартомы радужки являются па- тогномоничным симптомом нейро-фиброматоза-1, известны казуистические случаи выявления узлов Лиша на одном глазу у пациента с нейрофибро-матозом-2.
Ретинальные гамартомы при нейро-фиброматозе-2 клинически выглядят как комбинированные гамартомы пиг¬ментного эпителия и сетчатки. При морфологическом исследовании гамартомы у одного пациента с нейро-фиброматозом-2 обнаружены признаки интраретинальной глиальной пролиферации. Обычно комбинированные гамартомы пигментного эпителия и сетчатки бывают односторонними, но описаны и двусторонние поражения. При офтальмоскопии комбинированные гамартомы пигментного эпителия и сетчатки определяют в виде полиморфных слегка проминируюших бело-серых масс, вероятно, представляющих собой пролиферирующие ретинальные и эпиретинальные ткани, окруженные обширными темно-коричневыми зонами атрофии пигментного эпителия и хоровдеи (рис. 11.5).

Гамартомы обычно локализуются в макулярной или юкстапапиллярной области. В ряде случаев формирование гамартомы приводит к витреоретинальным тракциям, обусловливающим деформацию диска зрительного нерва и/или гетеротопию сосудов и сетчатки в пери фокальных областях. При флюоресцентной ангиографии у детей с комбинированными гамартомами пигментного эпителия и сетчатки могут быть нарушения проницаемости капилляров.
Жалобы на снижение зрения или диплопию предъявляют приблизительно 13 % пациентов с нейрофиброматоом-2. Острота зрения у больных с юкстапапиллярными или макулярными комбинированными гамартомами макулы значительно снижена и обычно варьирует в пределах от 0,01 до 0,2.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0