Факоматоз | Детская офтальмология


Содержание:
Описание
Факоматоз, относящийся к нейроэктодермальным расстройствам, представляет собой группу синдромов, при которых кожа, глаз и центральная нервная система вовлечены в процесс тканевой гиперплазии доброкачественного течения. В эту группу заболеваний входят нейрофиброматоз, туберозный склероз, болезнь Гиппеля-Линдау (Hippel-Lindau) и синдром Стерджа-Вебера (Sturge-Weber).↑ Нейрофиброматоз
Нейрофиброматоз делят на две аутосомно доминантных формы, характеризующихся различным клиническим течением: (1) нейрофиброматоз I типа (НФ1) — синдром Реклингхаузена (Recklinghausen) и (2) НФ2 — двусторонний акустический нейрофиброматоз. Описаны дополнительные формы нейрофиброматоза, включающие сегментарный нейрофиброматоз, кожный смешанный НФЗ, вариант НФ4 и НФ7 с поздним началом. Не выяснено, являются ли все представленные формы отдельными самостоятельными заболеваниями.
↑ НФ1
Диагностические критерии
Для установления диагноза НФ1 критериями были определены нижеследующие признаки. Необходимо наличие, по меньшей мере, двух из них, чтобы установить диагноз.
- Пять и более пигментных пятен цвета кофе с молоком» более 5 мм в диаметре, у ребенка препубертатного возраста и шесть или более пигментных пятен цвета кофе с молоком диаметром более 15 мм у больного в постпубертатном возрасте.
- Две и более нейрофибромы любого типа или одна плексиформная нейрофиброма.
- Подмышечные или паховые пятнышки.
- Глиома зрительного нерва.
- Два или более узелка Лиша (Lisch).
- Характерные дефекты костей (псевдоартрозы болыпеберцовой кости или дисплазия крыла сфеноидальной кости) (рис. 24.1).
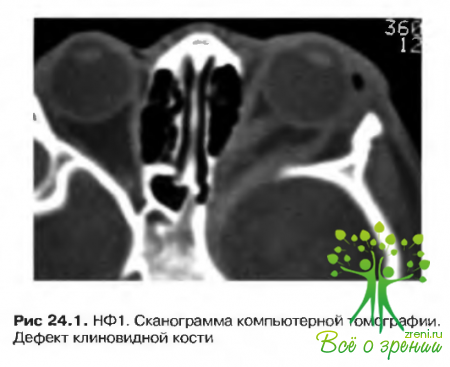
- Ближайшие родственники, страдающие НФ1.
Распространенность
Установлено, что распространенность нейрофиброматоза соответствует 1:3000-1:5000, таким образом, это заболевание является одним из наиболее часто встречающихся аутосомно-доминантных расстройств. Пенетрантность практически полная, в результате высокого уровня спонтанных мутаций у 50% больных нейрофиброматозом определяют новые мутации. Пораженный ген локализован на проксимальном длинном плече 17 хромосомы (17qll.2).
Общие проявления
- Пигментные пятна на теле цвета кофе с молоком. Однако эти изменения не являются патогномоничным признаком нейрофиброматоза и могут встречаться у здоровых людей.
- Мелкие пятнышки локализуются преимущественно в складках кожи — в подмышечных впадинах, в паховой области и под грудью у женщин.
Периферические нейрофибромы
Практически у всех больных с НФ1 к 16-летнему возрасту развиваются периферические нейрофибромы кожных покровов и, в более редких случаях, — нейрофибромы подкожной клетчатки, пальпируемые по ходу периферических нервов.
Плексиформные нейрофибромы
Своеобразные новообразования мягкой консистенции. Патогномоничный симптом для НФ1. Характерными признаками являются гипертрофия окружающих тканей, локальное разрастание ткани и гипертрихоз в пораженной зоне. При локализации процесса в орбите возможно значительное снижение зрения, обусловленное прямой компрессией зрительного нерва или амблиопией, развивающейся за счет птоза и/или косоглазия, спровоцированного опухолью.
Снижение способности к обучению
Хотя умственная отсталость при НФ1 встречается редко, может наблюдаться легкое снижение зрительного восприятия.
Офтальмологические проявления
Осмотр офтальмологом больного с подозрением на нейрофиброматоз важен не только для подтверждения диагноза, но и для выявления осложнении со стороны органа зрения и по возможности раннего назначения лечения.
Патологические изменения органа зрения могут локализоваться в орбите и включать:
- глиому зрительного нерва;
- оболочечную менингиому зрительного нерва;
- нейрофиброму орбиты;
- дефекты костей орбиты.
Экзофтальм может быть связан с изменением положения глазного яблока, впоследствии присоединяются косоглазие и амблиопия. Опухоли орбиты часто вызывают изменения зрительного нерва, проявляющиеся застойным соском, атрофией и, несколько реже, гипоплазией зрительного нерва.
Дополнительные изменения включают:
- оптоцилиарные шунты (особенно при метингиоме зрительного нерва);
- складчатость сосудистой оболочки;
- амавроз, определенный направлением взора. Неврологические исследования показаны для дифференциации различных причин экзофтальма при НФ1.
Веки
- Наиболее часто веки поражаются плексиформной нейрофибромой с характерной 5-образной деформацией края верхнего века. Косоглазие и/или птоз, возникающие в результате этих изменений, могут привести к появлению амблиопии.
- Врожденный птоз встречается даже при отсутствии опухоли орбиты.
Радужка
Узелки Лиша (меланоцитарные гамартомы радужки) патогномоничны для НФ1. При НФ2 они встречаются редко. С возрастом распространенность их при НФ1 увеличивается. В раннем детском возрасте узелки Лиша наблюдаются не часто, но к 20 годам обнаруживаются практически у 100% больных.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Зрительный нерв
Вовлечение в патологический процесс зрительного нерва проявляется глиомами зрительного нерва (астрогиномами) (рис. 24.2). 70% всех глиом зрительного нерва встречаются у больных с НФ1. Истинную частот}' их распространенности при НФ1 определить трудно в связи с отсутствием симптоматики и поэтому субклиническим течением. Приблизительно V 15% больных с НФ1 и нормальной остротой зрения радиографически выявляются глиомы зрительного нерва. Эти новообразования подразделяют на две категории.
Передние (глиомы орбиты)
Эти глиомы представлены экзофтальмом, потерей зрения и, изредка, изменением положения глазного яблока Вовлечение в процесс зрительного нерва выражается в его атрофии, дисплазии, прямом поражении опухолью и застойным соском. Иногда на стороне опухоли формируются оптоцилиарные сосудистые шунты. В результате изменений часто возникает косоглазие.
Задние (миомы хиазмы)
Симптомами этих глиом являются гидроцефалия, эндокринная патология и снижение зрения в сочетании с нистагмом. Нистагм может быть вертикальным, ротаторным или асимметричным (изредка имитирует спазмы нутане). Часто возникает диссоциированный вертикальный нистагм.
Конъюнктива
Нейрофибромы конъюнктивы наблюдаются редко и располагаются, как правило, в лимбальной зоне.
Роговица
При НФ1 встречается утолщение нервов роговицы, однако этот симптом не является патогномоничным. Значительно чаще это расстройство возникает при синдроме множественной эндокринной неоплазии.
Увеалъный тракт
Пигментные гамартомы сосудистой оболочки встречаются у 35% больных. Диффузные нейрофибромы вызывают утолщение всего увеального тракта, приводя к глаукоме.
Сетчатка
При НФ1 сетчатка редко вовлекается в патологическим процесс. Имеются единичные сообщения о случаях возникновения астроцитарных гамартом сетчатки и ее пигментного эпителия.
Исследования
- Компьютерная томография (КТ) или магнитно-резонансная томография (МРТ) мозга и орбиты позволяют обнаружить патологию костей, менингиомы н глиомы зрительного нерва.
- Зрительные вызванные потенциалы (ЗВП) оказывают помощь в оценке состояния хиазмы, а также при контроле за динамикой хназмальных глиом.
Тактика ведения
Тактика ведения больных с НФ1 до настоящего времени остается дискутабельнон. Лечение, как правило, комплексное. Неотъемлемым компонентом в общем комплексе назначений является генетическая консультация всех членов семьи больного.
Лечение осложнений нейрофиброматоза со стороны органа зрения сложное и включает:
Плексиформная нейрома
Химио- и лучевая терапия неэффективны. Хирургическое удаление технически сложно и имеет высокий уровень осложнений. При подозрении на амблиопию рекомендуют проведение окклюзии.
Глиомы зрительного нерва и хиазмы
Вопросы лечения по сей день сохраняют свою актуальность. Консервативное лечение показано при опухолях маленького размера и сохранных зрительных функциях. У пациентов старшего возраста иногда рекомендуют лучевую терапию (у детей младшей возрастной группы лучевая терапия может осложниться отставанием в умственном развитии).
Хирургическое вмешательство целесообразно на слепом глазу при выраженном экзофтальме. Хирургическое удаление глиом хиазмы технически невозможно. В некоторых случаях удаляют лишь сопутствующие кисты. Все более популярной в лечении хиазмальных глиом при их сочетании с дисфункцией гипоталамуса становится химиотерапия. Некоторым больным с глиомами хиазмы при повышении внутричерепного давления показано шунтирование. Желательно проведение эндокринологического обследования всех больных с глиомами хиазмы.
Глаукома
Обычно требует хирургического вмешательства, при этом имеет неблагоприятный прогноз.
НФ2 (двусторонний акустический) (рис. 24.2)
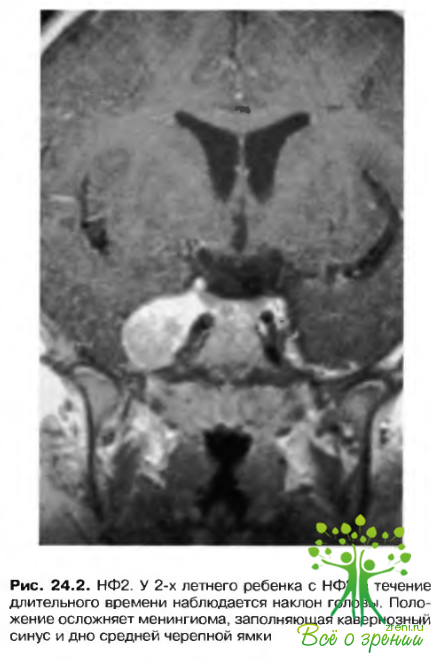
Эта форма заболевания менее распространена, чем НФ1. Ответственный за возникновение расстройства ген локализован около центра длинного плеча 22 хромосомы.
Общие проявления
- Пятна на теле цвета кофе с молоком, возникают приблизительно V 60% больных.
- Нейрофибромы кожи наблюдаются приблизительно в 30% случаев. Плексиформные фибромы встречаются редко.
Проявления со стороны центральной нервной системы
Отличительным признаком нейрофиброматоза являются двусторонние невриномы слухового нерва. Остальные черепно-мозговые нервы могут также вовлекаться в процесс по мере роста опухоли, особенно V, VI и VII пара. Распространены глиомы, менингиомы и шваиномы.
Проявления со стороны органа зрения
- Узелки Лита, если встречаются, то редко.
- Задняя субкапсулярная катаракта возникает часто, но мало влияет на остроту зрения.
- Комбинированные гамартомы пигментного эпителия и сетчатки.
- Эпиретинальные мембраны с легким снижением зрения.
В большинстве случаев глазных проявлении НФ2 лечения не требуется. Необходимость в лечении возникает при появлении двусторонней менингиомы VIII пары черепно-мозговых нервов, особенно если опухоль имеет маленький размер.
↑ Туберозный склероз
Характерными проявлениями туберозного склероза считают триаду симптомов — умственную отсталость, эпилептические припадки и везикуло-папиллярные кожные высыпания в сочетании с опухолями головного мозга. Туберозному склерозу сопутствует развитие множественных опухолей. Заболевание наследуется по аутосомно-доминантному типу. Однако по крайней мере в 75% случаев эта патология обязана своим происхождением спонтанным мутациям. Полный синдром никогда не проявляется в двух последовательных поколениях. Заболевание сопровождается по меньшей мере двумя различными местоположениями гена — в 9 хромосоме (9д34) и 11 хромосоме (11ц22-ц23).
Диагностические критерии
Диагноз туберозного склероза может быть поставлен, если присутствует один основной его признак или два второстепенных.
Основные признаки
- Околоногтевая фиброма (рис. 24.3).

- Гамартомы сетчатки (более одной).
- Ангиофибромы лица (рис. 24.4).

- Субэпендпмальные глиальные узелки (выявляются при визуализации неврологических исследований).
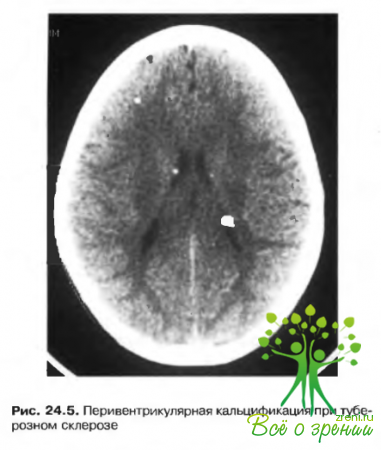
- Множественные бугорки в коре головного мозга (рис. 24.5).
- Двусторонние ангиомиолиномы почек.


Диагноз туберозного склероза может быть также установлен при наличии у больного одного из ниже перечисленных признаков:
- шагреневое утолщение кожи;
- фиброзные бляшки в области лба;
- множественные карднальные рабдомиомы;
- гигантоклеточная астроцитома;
- изолированная факома сетчатки;
- изолированный бугорок в коре головного мозга.
Второстепенные признаки
Если признаки первого порядка отсутствуют, но у больного обнаружено появление двух нижеперечисленных отличительных черт, то может быть выставлен диагноз туберозного склероза:
- депигментированные пятна;
- двусторонний поликистоз почек;
- кардиальная рабдомиома;
- ангиомиолипома почки;
- лимфоангиомиоматоз легких;
- распространенная гипомнелинизация коры и подкорки.
Общие характеристики
Изменения кожных покровов
Ангиофиброматоз
Редко возникает до 2-х летнего возраста, но в периоде полового созревания наблюдается с частотой 85%. Патогномонично расположение повреждения в виде бабочки на коже носощечной области, не затрагивая верхнюю губу, но вовлекая в процесс подбородок.
Гипопигментированные пятна
Наиболее часто принимают форму «пепельной листвы» и имеют вид белесых пятен продолговатой формы, обычно по линии дерматома. Могут возникать уже на первом году жизни (чтобы их увидеть может потребоваться ультрафиолетовая лампа Wood). Симптом не рассматривают как натогномоничный признак туберозного склероза.
Шагреневые пятна
У 40% больных туберозным склерозом обнаруживают депигментированные утолщения кожи, обычно локализованные на поясничной области (рис. 24.6).

Фиброзные бляшки в области лба
Этот патогномоничный симптом может наблюдаться уже при рождении, но в целом наблюдается лишь в 25% случаев. Бляшки имеют вид выпуклых красных «восковидных» новообразований кожных покровов.
Фибромы
Околоногтевые и подногтевые фибромы, исходящие от ногтей рук и ног. Менее распространены фибромы десны. На задней поверхности шеи и плечах возникают выросты кожи. Фибромы встречаются у 50% больных туберозным склерозом.
Изменения со стороны внутренних органов
У больных туберозным склерозом в патологический процесс могут вовлекаться почки, сердце, легкие и другие внутренние органы.
Почки
Множественные ангпомнолиномы н доброкачественные кисты почек.
Сердце
Рабдомиомы, обычно множественные, но, как правило, имеющие бессимптомное течение.
Легкие
Субплевральные кисты, фиброз и вторичная эмфизема легких.
Поражение нервной системы
- Припадки:
- а. в период новорожденности и в раннем детстве — инфантильные судороги;
- б. у детей старшего возраста — по типу grand mal.
- а. в период новорожденности и в раннем детстве — инфантильные судороги;
- Умственная отсталость наблюдается приблизительно у 60% больных.
- Внутричерепные повреждения.
- Формирование новообразований в коре и подкорке головного мозга. Новообразования представляют собой гамартомы и выявляются при КТ и МРТ. В некоторых случаях возникает малигнизация, являющаяся причиной 25°о случаев ранней смерти при туберозном склерозе.
Офтальмологические проявления
Наиболее характерными проявлениями туберозного склероза со стороны органа зрения являются гамартомы сетчатки и другие повреждения заднего отрезка глаза.
Факома (гамартома сетчатки)
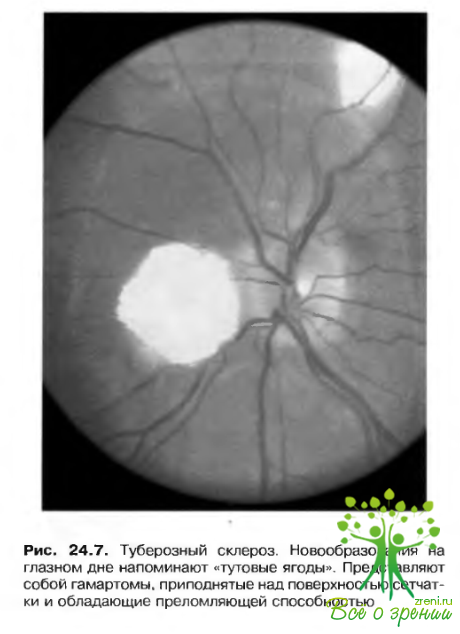
Встречается приблизительно у 50% больных и в 50% случаев является двусторонней. Может выглядеть как плоское, гладкое и полупрозрачное образование, а может иметь вид приподнятой, обызвествленной «тутовой ягоды». Наблюдаются и комбинированные формы, объединяющие черты обоих типов. Эти повреждения. как правило, не склонны к выраженному росту (рис. 24.7).
Сосудистые изменения сетчатки
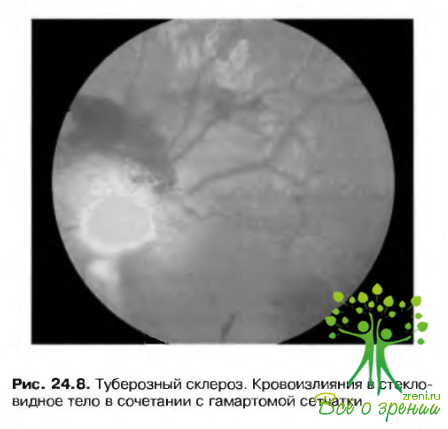
Сопутствуют опухолям. Характерны аневризмы сосудов и пороки развития, которые могут стать причиной кровоизлияний в стекловидное тело (рис. 24.8).
Патология пигментного эпителия
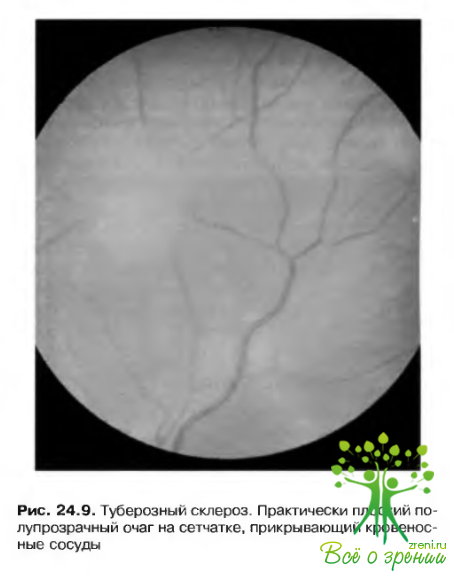
Описаны изменения пигментного эпителия, возникающие на средней периферии сетчатки п создающие картину «перфорированной» поверхности за счет появления зон депигментации (рис. 24.9).
Патология диска зрительного нерва
Могут наблюдаться отек диска зрительного нерва или атрофия зрительного нерва. Изредка встречаются косое положение диска зрительного нерва и колобомы.
Экстраретинальные расстройства
Описаны и другие особенности состояния органа зрения при туберозном склерозе, включая:
- мегалокорнеа;
- кератоконус;
- задний эмбриотоксон;
- глаукому;
- катаракту.
↑ Синдром Стерджа-Вебера
Синдром Стерджа-Вебера редкое заболевание с вовлечением в патологический процесс нервной системы, органа зрения и кожи. В классической форме синдром проявляется багрово-красным пятном, локализованным па одной стороне лица в зоне иннервации тройничного нерва. Неврологическая симптома тика включает эпилепсию, умственную отсталость и гемниарезы.
Основным проявлением заболевания со стороны органа зрения является глаукома. Генетический механизм этого синдрома до конца не выяснен. Большинство авторов относят заболевание к соматической мутации в период раннего внутриутробного развития, но есть точка зрения, что причиной расстройства является летальный ген, сохранившийся за счет мозанчности.
Эмбриогенез синдрома, по-видимому, включает неполную дифференцировку первичной мозенхимы сосудов краниальной эктодермы и подлежащей нервной трубки в сроки от А до 8 недель гестационного возраста. С другой стороны, существует предположение, что дефект является результатом нарушения сообщения между кортикальными венами и верхним сагиттальным синусом, что обусловливает появление порока развития сосудов мягкой п паутинной оболочек мозга п формирование коллатеральной циркуляции.
Диагностические критерии
Синдром Стерджа-Вебера с классической триадой симптомов
- Нейро-дермалыный ангиоматоз.
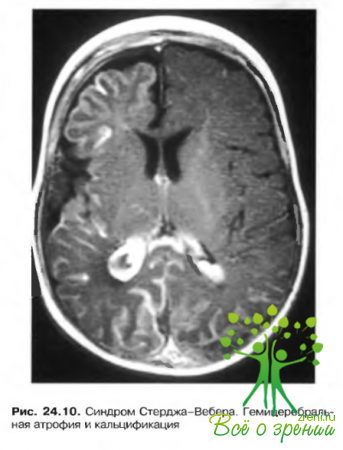
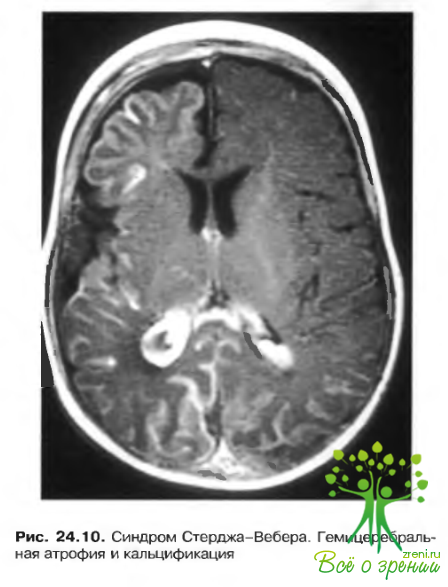
- Ангиоматоз мягкой и паутинной оболочек мозга (рис. 24.10).
- Патология органа зрения.
- Ангпоматоз кожных покровов.
Абортивная двусимптомная форма синдрома Стерджа-Вебера (рис. 24.11)

- Ангиоматоз органа зрения и кожных покровов.
- Нейро-дермальный ангиоматоз.
Абортивная моносимитомная форма синдрома Стерджа-Вебера
- Изолированный ангиоматоз мягкой и паутинной оболочек мозга.
- Изолированный ангиоматоз кожи в зоне иннервации тройничного нерва.
Расширенный синдром Стерджа-Вебера
Эти случаи включают сочетания с другими нейроэктодермальными синдромами:
- Синдром Клипнеля-Треноне-Вебера;
- окуло-дермальный меланоз (невус Ота [Ota]);
- нейрофиброматоз;
- туберозный склероз;
- болезнь Гиппеля—Линдау;
- болезнь Вибурна-Масона;
- мраморная кожа;
- нейродермальный меланоз;
- пигментоваскулярный факоматоз IVa типа.
↑ Болезнь Гиппеля-Линдау
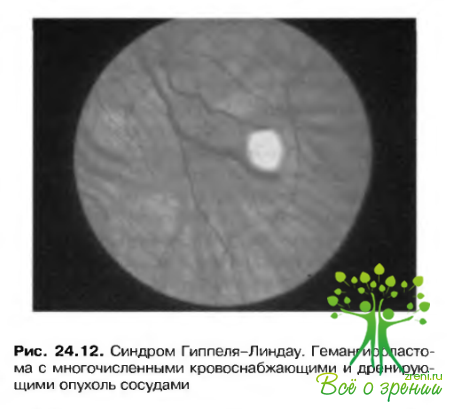
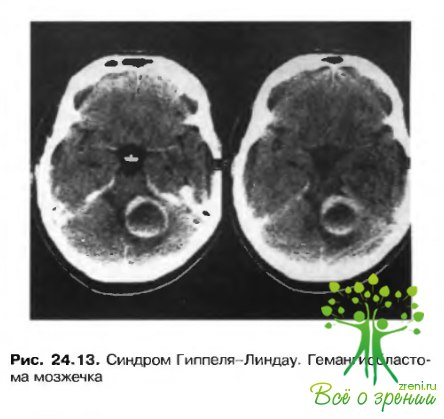
Ангноматоз сетчатки и мозжечка составляют синдром известный как болезнь Гиппеля-Линдау (рис. 24.12). Заболевание наследуется по аутосомно-доминантному типу. Ответственный за возникновение синдрома ген локализован на коротком плече 3 хромосомы (Зр25-р26). Основные признаки заболевания включают ангноматоз сетчатки, гемангиобластомы мозжечка, головного и спинного мозга (рис. 24.13). а также почечные карциномы п феохромоцитомы.
Характерной чертой этого расстройства является разнообразие клинической симптоматики. У одного больного редко обнаруживаются все патологические признаки заболевания.
Общие проявления
Неврологические
Повреждения центральной нервной системы практически всегда локализуются ниже намета мозжечка. Наиболее распространена гемангпобластома мозжечка, встречающаяся с частотой около 20%. Аналогичные повреждения возникают в головном н спинном мозге, но более редко. Могут наблюдаться спрингобульбия и сирингомнелия.
Поражения внутренних органов
Почки вовлекаются в патологический процесс с образованием карцином или гемангиобластом из паренхиматозных клеток. Реже возникает гемангиома поджелудочной железы. Феохромоци- гомы наблюдаются приблизительно у 10% больных. Параганглпома эпидидимуса не характерна для синдрома Гиппеля-Линдау.
Офтальмологические проявления
Приблизительно в 2/3 всех случаев заболевания наблюдается ангноматоз сетчатки, локализующийся, как правило, па средней периферии.
Описаны пять стадий развития этого повреждения.
- Стадия 1. Доклиническая; начальные скопления капилляров, незначительное их расширение по типу диабетических микроаневризм.
- Стадия 2. Классическая; образование типичных ангиом сетчатки.
- Стадия 3. Экссудативная; обусловлена повышенной проницаемостью сосудистых стенок ангиоматозных узлов.
- Стадия 4. Отслойка сетчатки экссудативного или тракционного характера.
- Стадия 5. Терминальная стадия; отслойка сетчатки, увеит, глаукома, фтизис глазного яблока.
Лечение, начатое на ранних стадиях развития заболевания, сопряжено с меньшим риском осложнений. Возможно проведение крио-, лазер- и лучевой терапии, а также хирургическая резекция.
Необходим скрининг больных синдромом Гиппеля-Линдау, включающий следующие исследования:
- ежегодный осмотр с регистрацией полученных данных;
- офтальмологическое обследование каждые 6-12 месяцев начиная с 6-летнего возраста;
- по меньшей мере однократное исследование мочи на наличие феохромоцитомы и повторение исследования при повышении или нестабильности кровяного давления;
- двусторонняя селективная ангиография почек по достижении больным 15-20-летнего возраста, с повторением процедуры каждые 1-5 лет;
- МРТ задней черепной ямки;
- Компьютерная томография поджелудочной железы и почек по достижении больным 15-20-летнего возраста, с повторением процедуры каждые 1-5 лет или внепланово при появлении соответствующей симптоматики.
Целесообразно проведение скрининга детей от пораженных синдромом родителей и/или других близких родственников с высоким риском появления заболевания.
Исследование проводят по следующим направлениям:
- осмотр с регистрацией полученных данных по достижении ребенком 10-летнего возраста;
- ежегодное офтальмологическое обследование начиная с 6-летнего возраста или при появлении подозрительной симптоматики;
- по меньшей мере однократное проведение исследования мочи на наличие феохромоцитомы и повторение исследования при повышении или нестабильности кровяного давления;
- МРТ задней черепной ямки, КТ поджелудочной железы и почек по достижении 20-летнего возраста;
- эхография поджелудочной железы и почек после 15-20-летнего возраста;при возможности, исследование семейного анамнеза для выяснения, кто из членов семьи является носителем ответственного за возникновение синдрома гена.

Комментариев 0