Наследственная оптическая нейропатия Лебера


Описание
Оптическая нейропатия Лебера (атрофия зрительного нерва Лебера) — наследственное заболевание, характеризующееся быстро или постепенно развивающимися двусторонними нарушениями центрального зрения у соматически здоровых молодых людей.История исследования проблемы. Заболевание впервые описано Theodor Leber в 1871 г., который сообщил о 15 пациентах из четырех семей. В последующем многие авторы опубликовали свои наблюдения за семьями из Европы, Азии, Америки, Африки и Австралии, у членов которых в нескольких поколениях выявляли оптическую нейропатию.
Внимание большинства исследователей было сосредоточено на изучении механизмов наследования патологии. Хитросплетения проблемы исследования механизмов наследования оптической нейропатии Лебера заключаются в том, что заболевание передается исключительно по материнской линии и развивается преимущественно у лиц мужскою пола. В 1963 г. A. van Senus доказал, что оно никогда не передается мужчинами. В то же время при Х-сцепленных рецессивных заболеваниях ожидаемая частота поражения женщин не превышает 1 %, что значительно ниже уровня, наблюдаемого при оптической нейропатии Лебера. Основываясь на этих противоречивых фактах, D.C. Wallace (1970) сделал следующее заключение: «Поверхностное изучение пораженной семьи создает впечатление о сцепленном с полом наследовании. Исследователь, завершающий работу, может предполагать цитоплазматическое наследование. Более детальный анализ приводит к путанице». В своих последующих работах он подчеркнул важное значение внехромосомных материнских факторов. Некоторые авторы не исключали возможности переноса персистирующих вирусоподобных субстанций в материнскую овоплазму или трансплацентарного поражения эмбриона.
Одновременно изучалось влияние факторов внешней среды на течение заболевания. J. Wilson (1965) обнаружил корреляцию между тяжестью клинических проявлений оптической нейропатии Лебера и курением. На основании своих наблюдений он предположил, что в основе зрительных расстройств у пациентов с оптической нейропатией Лебера лежат нарушения метаболизма цианидов. Позднее ТА. Berninger и соавт. (1989) выявили повышение уровня цианидов в крови у пациентов с оптической нейропатией в острой фазе. К. Tsao и соавт. (1999) подтвердили корреляцию стажа и объема курения с пенетрантностью и экспрессивностью оптической нейропатии Лебера.
Генетические исследования. В настоящее время доказано, что развитие нейропатии Лебера обусловлено точковыми мутациями в митохондриальной ДНК, приводящими к замене одной аминокислоты другой. Предпола¬гаемое митохондриальное наследование оптической нейропатии Лебера было подтверждено в 1988 г. D.C Wallace и соавт., которые первыми идентифицировали точку мутации митоховдриальной ДНК в нуклеотвдной позиции 11778 у больных с оптической нейропатией Лебера из девяти семей. В результате этой мутации происходит замещение аргинина на гистидин в кодоне 340 гена субъединицы 4 НАДФ-дегидрогеназы. Мутация 11778 идентифицирована в 50—60 % всех обследованных генетиками семей, члены которых были больны нейропатией Лебера.
Другую патогенетически значимую (первичную) мутацию в положении 3460 митохондриальной ДНК у больных с нейропатией Лебера из трех семей, у членов которых не выявлена мутация 11778, установили К. Huoponen и соавт. (1991). Данная мутация в колоне 52 гена, кодирующего субъединицу 1 НАДФ-дегидрогеназы, приводит к замене аланина треонином. Позднее N. Howell и соавт. (1991), а также D.R. Johns (1992) идентифицировали мутацию в нуклеотидной позиции 3460 еще в 15 семьях больных с оптической нейропатией Лебера. Мутация в точке 3460 определена приблизительно у 8 % пациентов с нейропатией Лебера.
Вскоре бьыи обнаружены мутации в нуклеотидных позициях 15257 и 15182 в гене апоцитохрома b. При обследовании 120 семей, члены которых были больны оптической нейропатией Лебера, мутация 15257 выявлена у 8 % родословных.
D.Besch и соавт. (1999) установили у больного с оптической нейропатией Лебера, его матери и четырех непораженных членов этой семьи по материнской линии первичную точковую мутацию митохондриальной ДНК в нуклеотидной позиции 14568. Эта мутация обусловливала замену глицина на серии в гене ND6. У матери пробанда не обнаружено зрительных расстройств, но при офтальмоскопии выявлена перипапиллярная микроангиопатия.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Это далеко не весь перечень мутаций, установленных у больных с оптической нейропатией Лебера. В настоящее время известно около 20 точковых мутаций. Во всех случаях они затрагивают компоненты комплексов ( и III дыхательной цепи окислительного фосфорилирования.
Комплекс 1 (NADH-дегидрогеназный комплекс) — самый крупный белковый компонент дыхательной цепи, содержащий более 22 полипептидных цепей, кодируемых как митохондриальным, так и ядерным геномами. Установлено, что мутация G3460A в гене ND1 в культивированных фибробластах приводит к снижению активности NADH-дегидрогеназы до 40 % по сравнению с нормой, при этом связанный с комплексом I синтез АТФ остается неизменным.
V.Carelli и соавт. (1999) исследовали влияние мутаций 14459 и 14484 в субьединице ND6 (Met 64 Val) на комплекс I. Мутация 14459 выявлена у больных с оптической нейропатией Лебера, сочетающейся с мышечной дистонией. Данная мутация индуцирует восстановление специфической активности NADH-дегидрогеназного комплекса и увеличивает его чувствительность к децилубихинолу — субстрату комплекса Ь-С| дыхательной цепи (комплекс II). Таким образом, комплекс I ингибируется продуктом катализируемой им реакции. Мутация 14484 не оказывает подобного влияния на специфическую активность комплекса I, однако обусловливает более высокую чувствительность этого ферментного комплекса по сравнению с немутированными формами к ингибиторам — аналогам убихинона.
Анализ 70 различных последовательностей ND6-субъединицы показал, что мутации 14484 подвержена наиболее консервативная область белковой молекулы, локально напоминающая участок цитохрома Ь, взаимодействующий с убихиноном или убихинолом в цитохромоксидазном комплексе (комплекс III).
Таким образом, все мутации воздействуют на систему окислительного фосфорилирования, нарушая тем самым энергетический обмен в клетке. Согласно гипотезе A. Sadun (1998), в результате уменьшения числа молекул АТФ ниже определенного порогового уровня блокируется антероградный аксональный транспорт митохондрий, что приводит к дефициту АТФ и в итоге к гибели нейрона.
Идентификация диагноза с помощью анализа митохондриальной ДНК показала, что оптическая нейропатия Лебера охватывает более широкий возрастной диапазон, а женщины болеют значительно чаще, чем предполагали раньше. Н. Thicme и соавт. (1999) сообщили о семье, у 9 членов которой была оптическая нейропатия Лебера, обусловленная мутацией 11778 субъединицы ND4, восемь из них — девочки в возрасте до 10 лет.
Пять мутаций (11778, 3460, 14484, 15257 и 14568) вместе составляют свыше 90 % точковых мутаций, выявленных во всех родословных с фенотипом оптической нейропатии Лебера. Предполагают, что каждая из них имеет первичное патогенетическое значение. Идентифицированы также несколько вторичных мутаций, которые, воздействуя сочетанию как синергисты, могут вызывать развитие оптической нейропатии Лебера.
R.I Oostra и соавт. (1994) исследовали распределение 7 мутаций митохондриальной ДНК и их связь с клиническими проявлениями оптической нейропатии Лебера у 334 пациентов из 29 семей. Мутации в нуклеотидных позициях 11778, 3460 и 14484, встречающиеся только при оптической нейропатии Лебера, были обнаружены в 15, 2 и 9 семьях соответственно. В 3 семьях не выявлена ни одна из этих мутаций. Мутации в нуклеотидных позициях 15257, 13708, 4917 и 4216, которые ранее были обнаружены как у пациентов с оптической нейропатией Лебера, так и у здоровых людей, идентифицированы в 1, 10, 3 и 12 семьях соответственно. Комбинации мутаций митохондриальной ДНК обнаружены у членов большинства обследованных семей. У пациентов из 11 семей, в которых была выявлена только одна мутация — 11778, заболевание манифестировало в среднем в возрасте 29,2 лет, а итоговая острота зрения составляла в среднем 0,113. Тяжесть фенотипических проявлений оптической нейропатии Лебера у больных с мутациями в других нуклеотидных позициях зависела от митохондриального генотипа.
В семьях больных с оптической нейропатией Лебера описано явление гетероплазмии: количество мутантной митоховдриальной ДНК варьирует у разных пациентов от 5 до 300 % от всей имеющейся митоховдриальной ДНК. Заболевание развивается в тех случаях, когда процентное содержание мутантной ДНК достигает порогового значения. Количество мутантной ДНК в клетках организма коррелирует с тяжестью клинических симптомов. В процессе эволюции гетероплазмия митохондриальной ДНК может эффективно поддерживаться на протяжении нескольких поколений. При этом у части лиц возможна фиксация одной из форм митохондриальной ДНК в результате случайной полной элиминации другой формы в ходе оогенеза. Так, в некоторых случаях благодаря гетероплазмии у матери мутантная ДНК не передается детям. М.Т. Lott и соавт. (1990) сообщили о родословных, в которых количество мутантной ДНК увеличивалось от поколения к поколению, коррелируя с тяжестью фенотипических проявлений. Авторы также обнаружили, что содержание мутантной ДНК в крови и волосах различно у одного и того же субъекта. Таким образом, выявление и количественная оценка гетероплазмии имеет чрезвычайно важное значение для диагностики, прогнозирования и определения фенотипической экспрессии.
Хотя атрофию зрительного нерва Лебера традиционно считают семейным наследственным заболеванием, в настоящее время достаточно часто клиницисты наблюдают больных, манифестацию заболевания у которых можно рассматривать как спорадическую. Семейные случаи оптической нейропатии Лебера составляют 43 % от общего числа пациентов для мутации 11778, 78 % - для мутации 3460, 65 % — для мутации 14484 и 57 % — для мутации 15257.
Клинические проявления. Оптическая нейропатия Лебера развивается у соматически здоровых людей, как правило, в возрасте 18—30 лет, однако известны случаи развития заболевания (мутация 11778) у 8-летнего ребенка, а также у мужчины в возрасте 73 лет. P.Pezzi и соавт. (1998) сообщили о 10-летнем итальянце с оптической нейропатией Лебера, обусловленной мутацией N01/3460. В Европе, Австралии и США мужчины составляют 80—90 % больных.
Заболевание проявляется острым или подострим снижением остроты зрения, появлением центральной скотомы в поле зрения. Потеря зрения может быть внезапной, а может начинаться постепенно и прогрессировать в течение 2 лет и более. Заболевание двустороннее, но в большинстве случаев поражение правого и левого глаз развивается асинхронно. Нарушения центрального зрения парного глаза при неодновременном поражении появляются в среднем через 2 мес. При офтальмоскопии могут быть выявлены псевдоотек и проминенция диска зрительного нерва, телеангиэктатические расширения капилляров пре- и перипапиллярной сети, извитость сосудов (рис. 14.3). В дальнейшем у части пациентов происходит побледнение височной половины диска зрительного нерва. Описанные выше изменения на глазном дне выявлены у 58 % пациентов с мутацией 11778.

Флюоресцентная ангиография. ФАГ при оптической нейропатии Лебера малоинформативна и ее используют только для дифференциальной диагностики с оптическим невритом, застойным диском, передней ишемической оптической нейропатией или псевдоотеком диска зрительного нерва. При ФАГ у пациентов с оптической нейропатией Лебера не определяется гиперфлюоресценция диска и папиллярных сосудов (см, рис. 14.3).
Зрительные функции. Острота зрения. Итоговая острота зрения зависит от типа мутации и варьирует от 0,8 до 0,01. В наиболее тяжелых случаях у больных с мутацией 11778 зрение снижается до 0. В то же время у больных с мутацией 3460 финальная острота зрения в неблагоприятных случаях равна «правильной светопроекции», а у больных с мутацией 15257 — «движению руки у лица». У пациентов с мутацией 14484 при плохом исходе сохраняется возможность счета пальцев. Раньше считали, что в тех случаях, когда заболевание манифестирует в возрасте до 15 лет, прогноз в отношении зрения более благоприятный, но в последующих исследованиях этой тенденции не отмечено.
Острота зрения у некоторых больных с оптической нейропатией Лебера может частично восстанавливаться, что определяется главным образом типом мутации и в меньшей степени зависит от внешних факторов, в частности от злоупотребления курением и алкоголем. Например, острота зрения незначительно повышается лишь у 4 % больных с мутацией 11778 в среднем через 36 мес после начала заболевания, тогда как из больных с мутацией 3460 — у 22 % в среднем через 68 мес, из пациентов с мутацией 15257 — у 28 % через 16 мес, а из пациентов с мутацией 14484 — у 37 % через 16 мес. Y. Hotta и соавт. (1993) описали случай атипичного течения оптической нейропатии Лебера с мутацией 11778 с асимметричным поражением и неожидан но высокой конечной остротой зрения 0,8 для OD и 0,05 для OS. М. Nakamura и М. Yamamoto (2000) наблюдали подростка, у которого заболевание, обусловленное мутацией 11778, манифестировало в возрасте 15лет, сопровождаясь снижением остроты зрения до 0,1/0,1. В дальнейшем было отмечено быстрое повышение остроты зрения до 0,6/0,7, которая не изменялась в течение последующих 4 лет наблюдения. P.Pezzi и соавт. (1998) сообщили о 10-летнем мальчике с оптической нейропатией Лебера (мутация ND1/3460), у которого происходило постепенное асинхронное снижение остроты зрения обоих глаз до 20/300, сочетавшееся с билатеральной цекоцентральной скотомой и дисхроматопсией. В последующие 2 года острота зрения каждого из глаз постепенно восстановилась до 1,0 и оставалась неизменной в течение 9 лет наблюдения.
Обнаружение у больного с оптической нейропатией Лебера мутации в позиции 11778 является неблагоприятным прогностическим признаком в отношении зрения. У больных с мутацией 3460, наоборот, относительно оптимистичный зрительный прогноз: приблизительно у 20 % из них острота зрения частично восстанавливается (иногда до 0,7—0,8). В то же время в семьях, где выявлена мутация 3460, по сравнению с семьями, в которых определяется мутация 11778, болеет оптической нейропатией Лебера значительно чаще не один член семьи, а несколько.
Поле зрения. В ранней стадии заболевания в поле зрения выявляют относительную центроцекальную скотому. Позднее формируется обширная (до 10— 15°) центральная абсолютная скотома, которая почти у всех больных остается до конца жизни. Довольно часто наблюдается распространение центральной скотомы к периферии вверх или вниз.
В некоторых случаях возможна частичная или полная регрессия изменений в поле зрения. В период восстанов¬ления зрительных функций у пациентов с оптической нейропатией Лебера положительная динамика изменений в поле зрения обычно предшествует улучшению остроты зрения и цветового зрения. Как правило, значительное повышение остроты зрения отмечают у тех больных, у которых в пределах обширной центральной скотомы появляются небольшие «островки» восстановленной яркости ой чувствительности. До тех пор, пока «фенестрации» скотомы не происходит, острота зрения у пациентов с оптической нейропатией Лебера не повышается.
Цветовое зрение. У большинства больных с оптической нейропатией Лебера выявляют нарушения цветового зрения в красно-зеленой части спектра, как при приобретенных поражениях зрительного нерва. Имеются сообщения о нарушениях цветового зрения у носителей, преимущественно по типу тританопии.
Пространственная контрастная чувствительность. В начальной фазе заболевания определяют снижение контрастной чувствительности в области средних и высоких пространственных частот. По мере снижения зрения нарушения пространственной контрастной чувствительности распространяются на весь частотный диапазон.
Электрофизиологические исследования. ЭРГ и ЭОГ у пациентов с оптической нейропатией Лебера не изменяются.
При регистрации паттерн-ЭРГ выявляют значительное снижение амплитуды компонента N95, который в некоторых случаях не удается выделить из фоновой активности. Амплитуда компонента Р50, как правило, не изменяется.
При регистрации ЗВП на вспышку отмечается снижение амплитуды компонента Р100 и удлинение его латентности. Некоторые авторы подчеркивают, что у больных с нейропатией Лебеpa основной позитивный компонент Р100 ЗВП раздваивается, образуя так называемый ПНП-комплекс. Данный электрофизиологический феномен не является патогномоничным для оптической нейропатии Лебера, так как наблюдается и при поражениях зрительных путей другой этиологии.
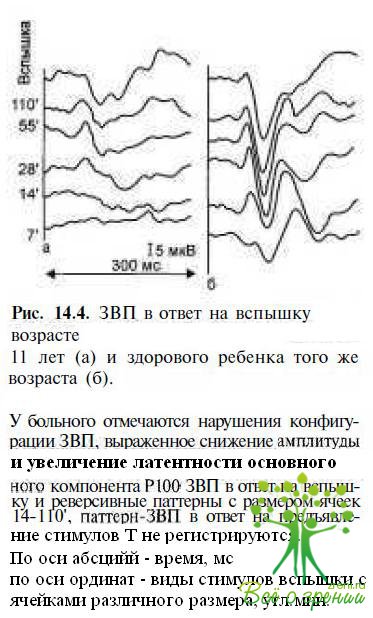
Паттерн-реверсивные ЗВП резко снижены по амплитуде, конфигурация их грубо нарушена (невозможно определить некоторые компоненты ответов), латентность компонента Р100 значительно увеличена (рис. 14.4). В начальной фазе заболевания изменяются преимущественно ЗВП, регистрируемые на паттерны с ячейками размером 55—7' (средние и высокие пространственные частоты), позднее выявляют нарушения ответов и на стимулы 220—110' (низкие пространственные частоты). У пациентов с остротой зрения ниже 0,01 паттерн-реверсивные ЗВП не регистрируются.
Системные проявления. У больных с оптической нейропатией Лебера и их родственников по материнской линии достаточно часто выявляют системные нарушения. В ряде родословных обнаружены различные кардиологические отклонения. В 9 % финских семей, где есть больные с оптической нейропати- ей Лебера, часто диагностируют синдромы преждевременного возбуждения миокарда, например синдромы Вольфа — Паркинсона — Уайта или Лоуна — Гаи она — Левина. Аналогичные отклонения обнаружили Y- Mashima и соавт. (1996) у 8 % пациентов с оптической нейропатией Лебера и различными типами мутаций митохондриальной ДНК из 35 японских семей. У членов афроамериканской родословной с мутацией 11778 было установлено удлинение интервала Q— Т на ЭКГ. Описаны также аномалии скелета и различные неврологические симптомы (атаксия, нарушение сухожильных рефлексов, сенсорные нейропатии).
В последнее десятилетие появились сообщения о сочетании у некоторых пациентов оптической нейропатии Лебера и рассеянного склероза. Авторы этих исследований не исключали возможную патогенетическую связь между рассеянным склерозом и оптической нейропатией Лебера. Определенный гаплотип митохоцдриальной ДНК выявлен только у больных с рассеянным склерозом, сочетающимся с оптическим невритом, но не обнаружен у пациентов с рассеянным склерозом без сопутствующих зрительных расстройств. У 2 больных с рассеянным склерозом была выявлена мутация 15257 митохондриальной ДНК. М.Т. Bhatti и N.J. Newman (1999) сообщили о нескольких пациентах с демиелинизирующим поражением ЦНС, сочетавшимся с оптической нейропатией Лебера, обусловленной мутациями 11778 и 14484. В то же время U. Mayr-Wohlfart и соавт. (1996), а также D.S.Mojon и соавт. (1999) НС обнаружили первичных мутаций 11778, 3460 или 14484 ни у одного из 203 обследованных ими пациентов с рассеянным склерозом. Аналогичные результаты получили В. Kalman и соавт. (1998), а также Е. Wilichowski и соавт. (1998). У пациентов с рассеянным склерозом, дебютом которого был острый оптический неврит, выявлены только вторичные точковые мутации в нуклеотадных позициях 4917, 4216 и 13708. Таким образом, сочетание рассеянного склероза и оптической нейропатии Лебера встречается чрезвычайно редко и в настоящее время нет убедительных доказательств их патогенетической связи.
Лечение. В настоящее время не существует способов эффективной профилактики и лечения оптической нейропатии Лебера. Традиционно используемые стероиды, антагонисты цианидов, гидроксикобаламин не дают эффекта. Сомнительными кажутся и предлагавшиеся ранее хирургические методики. Придавая значение роли цианидов в патогенезе нейропатиии Лебера, I.Syme и соавт. (1983) применяли цистин внутрь по 4—8 г в день в сочетании с введением гидроксикобаламина внутримышечно по 1000 MKT 3 раза в неделю. У 35 % больных за период наблюдения, равный 1 году, острота зрения на фоне лечения повысилась до 0,3. Однако данное сообщение появилось в печати за несколько лет до того, как была установлена роль мутаций митохондриальной ДНК в патогенезе оптической нейропатии Лебера. Между тем опубликованные недавно результаты изучения особенностей естественного течения оптической нейропатии Лебера, обусловленной мутациями определенных типов, позволяют усомниться в эффективности предложенной схемы лечения. Например, в более поздних исследованиях была продемонстрирована возможность самопроизвольного восстановления зрения через 16 мес после начала заболевания у 37 % пациентов с оптической нейропатией Лебера, обусловленной мутацией 14484. Эти данные практически совпадают с эффектом лечения в исследовании, проведенном I.Syme и соавт. (1983).
Как и при других заболеваниях, сопровождающихся митохондриальными нарушениями, обоснованным представляется назначение коэнзима Q10 и АТФ, хотя выраженного терапевтического эффекта эти препараты не дали.
Статья из книги: Зрительные функции и их коррекция у детей | С.Э. Аветисов, Т.П. Кащенко, А.М. Шамшинова.

Комментариев 0