Болезни соединительной ткани | Патология органа зрения при болезнях обмена веществ


Описание
К настоящему времени описано множество глазных изменений при различных видах нарушений обмена веществ. Необходима- отметить, что иногда именно глазные симптомы являются наиболее ранними признаками патологии обменных процессов в организме.Своевременное выявление этих симптомов позволяет рано начать лечение и правильно выбрать метод для его осуществления. В связи с этим знание и умение обнаружить первые офтальмологические признаки является насущной необходимостью. Эта тем более важно, что диагностика болезней обмена у детей трудна ввиду их исключительного полиморфизма.
Нередко в пределах одной нозологической формы отмечаются разные клинические синдромы, неодинаковая тяжесть одних и тех же заболеваний - от стертых проявлений до тяжелых форм. Важно выявить характерные сочетания симптомов поражения органа зрения с признаками поражения других органов, так как нередко только по совокупности этих симптомов и динамике развития болезни удается в ранних стадиях установить правильный диагноз.
Изменения органа зрения при нарушениях обмена веществ обусловлены сложным процессом обмена в тканях глаза. Несовершество и незрелость ферментативных процессов у детей, отсутствие и недостаточность некоторых элементов, а также их накопление вызывают необратимые изменения в прозрачных средах глаза, сетчатке и других отделах глазного яблока.
Так, структура хрусталика и сетчатки, их метаболизм обусловливаются одновременным воздействием по крайней мере нескольких тысяч химических реакций, катализируемых различными ферментами. Основным источником энергии является глюкоза.
Нарушение обмена глюкозы приводит к накоплению органических кислот и других продуктов метаболизма, которые вызывают грубые изменения в тканях. Вследствие этого, например, при непереносимости галактозы отмечается катарактогенный эффект и появляются дегенеративные изменения в сетчатке.
Белки хрусталика органоспецифичны. Они синтезируются из аминокислот, которые проникают в него через капсулу хрусталика из влаги камеры.
В роговице отмечается большое количество кислых и нейтральных гликозаминогликанов, коллаген роговицы характеризуется очень высоким уровнем азота, глицина, пролина, оксипролина и небольшим количеством серосодержащих аминокислот и триптофана.
Для сетчатки как производного нервной системы характерны» сходные с другими нервными субстратами процессы метаболизма, поэтому она нередко изменяется при поражении мозга. Сетчатка отличается самой высокой активностью дыхания по сравнению с другими тканями организма.
Химическая энергия в виде АТФ в сетчатке образуется в основном за счет гликолиза и дыхания. Сетчатка, так же как и мозг, содержит большое количество глютаминовой кислоты, глютамина и микроэлементов, которые являются компонентом окислительных ферментов.
БОЛЕЗНИ СОЕДИНИТЕЛЬНОЙ ТКАНИ
Наиболее часто морфологической основой развития ряда наследственных болезней является поражение соединительной тканей. Наследственные болезни соединительной ткани мало изучены, дифференциальная диагностика их сложна, особенно у детей раннего возраста при приобретенных клинически сходных (фенотипические) заболеваниях. Для наследственных нарушений метаболизма соединительной ткани характерно поражение органа зрения.
Глазные симптомы могут быть ранними и единственными признаками наследственной патологии соединительной ткани, поэтому наряду с генеалогическими и биохимическими исследованиями при этих заболеваниях важное значение имеют офтальмологические методы диагностики.
Принято различать две основные группы болезней соединительной ткани: к первой группе относятся генетически обусловленные заболевания — мукополисахаридозы, синдром Марфана, гомоцистинурия и др., ко второй — болезни, в развитии которых определенную роль играют такие наследственные факторы, ревматизм, инфекционный неспецифический полиартрит, красная волчанка.
При патологических состояниях соединительной ткани первыми и самыми ранними признаками являются изменения в обмене кислых гликозаминогликанов, что свидетельствует о большой чувствительности этой ткани, в частности межуточного основного вещества, к обменным нарушениям в организме.
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
Химические свойства коллагена, в состав которого входят кислые гликозаминогликаны (КГАГ), обусловлены значительным количеством оксипролина, содержание которого особенно высоко в роговице и капсуле хрусталика, поэтому характер глазных изменений помогает также выявить некоторые стороны нарушений обмена КГАГ и оксипролина.
Особенно значительные изменения глаз отмечаются при наиболее тяжелой форме наследственного поражения соединительной ткани мукополисахаридозе. Заболевание называют также множественным дизостозом, липохондродистрофией, номедистрофией, остеохондродистрофией.
Термин «гаргоилизм» предложен из-за сходства больных с уродливыми скульптурными готическими фигурами. Эта группа наследственных заболеваний соединительной ткани характеризуется сочетанным поражением опорно-двигательного аппарата, внутренних органов, глаз и нервной системы. Заболевания протекают тяжело, диагностика их затруднена, особенно в ранних стадиях.
Наиболее тяжелым заболеванием является мукополисахаридоз I типа — синдром Гурлер, прототип всех мукополисахаридозов, характеризующийся прогрессирующим течением, а также тем, что больные умирают в ранние сроки (до 10—12 лет). Остальные шесть типов мукополисахаридоз а существенно отличаются от первого.
Мукополисахаридоз II типа - синдром Гунтера протекает менее тяжело. Ведущими симптомами являются изменения в костно- суставной системе, но менее выраженные, чем при I типе, а также снижение слуха.
Значительно отличается от первых двух типов мукополисахаридозов III тип - синдром Санфилиппо. Заболевание характеризуется наличием умственной отсталости на фоне незначительных изменений скелета и соматических нарушений.
В клинической картине мукополисахаридоза IV типа — синдрома Моркио на первое место выступают поражения костно-суставной системы деформации грудной клетки и черепа.
Мукополисахаридоз V типа — синдром Шейе — характеризуется почти полной сохранностью интеллекта, преимущественным поражением мелких суставов и умеренно выраженными соматическими изменениями.
Для мукополисахаридоза VI типа — синдрома Марото - Лами характерны грубые деформации скелета при сохранении нормального интеллекта.
Мукополисахаридозы всех типов, кроме синдрома Гунтера, наследуются по аутосомно-рецессивному типу. При синдроме Гунтера тип наследования рецессивный, сцепленный с полом, нередки семейные случаи заболевания.
Мукополисахаридоз VII типа обусловлен дефектом р-глюкуронидазы и по клиническим проявлениям напоминает мукополисахаридоз I типа, но без помутнения роговицы; тип наследования неизвестен.
Наиболее типичным симптомом поражения глаз при мукополисахаридозах является дистрофия роговицы. Наиболее рано помутнение роговицы развивается при синдроме Марото — Лами. Большинство исследователей не обнаруживают изменений глаз при синдроме Санфилиппо. Синдром Моркио также протекает с дистрофией роговицы, но она нередко появляется поздно, чаще у детей старше 10 лет. Дистрофию роговицы обнаруживают у 75— 90% больных мукополисахаридозами.
Из других симптомов при мукополисахаридозах описаны мегалокорнеа, гидрофтальм, атрофия и застой соска зрительного нерва, а также дегенерация сетчатки и глаукома.
При мукополисахаридозах проводят полное исследование глаза и его функций. Неврологические и генетические исследования выполняют по классической схеме.

Дифференциальную диагностику форм заболевания следует проводить на основе тяжести поражения органа зрения и соматических проявлений (табл. 31). У обследованных больных были выявлены следующие характерные общеклинические симптомы: увеличение и изменение формы головы, костно-суставные деформации.
Необходимо особо отметить, что мукополисахаридозы в начальных стадиях необходимо дифференцировать от рахита, гипотиреоза, инфекционного неспецифического полиартрита, полигландулярной недостаточности, липоидоза. Следует подчеркнуть также, что эти дети часто болеют простудными заболеваниями.
В дальнейшем у них выявляются грубые черты лица, и все больные отстают в росте. У них отмечаются пупочные и паховые грыжи, гепатоспленомегалия, пороки сердца. При всех формах мукополисахаридозов ранними признаками являются изменения глаз различной степени выраженности.
В процессе наблюдения за больными детьми выявлено, что наиболее ранние (у новорожденных и в первые месяцы после рождения) и тяжелые поражения глаз выявляются при мукополисахаридозе I типа. Они наблюдаются у всех больных.
У детей уже в первые месяцы жизни отмечаются длинные и густые ресницы, латеральный проптоз, пастозносгь век, особенно нижних. Конъюнктива век и глазного яблока отечная, цианотичная.
Наиболее выражен отек конъюнктивы по типу стекловидного на 3 и 9 часах у лимба по ходу кровеносных сосудов. Сосуды лимбальной области расширены, прорастают в прозрачные отделы роговицы. Кровоток в капиллярах замедлен, сосуды имеют форму крючков, местами наблюдается полный стаз.
Стенки сосудов утолщены. У всех больных выявляются макрокорнеа (12—13,5 мм) без расширения лимба и дистрофия роговицы различной степени выраженности. Помутнение роговицы, от нежного облаковидного до грубого диффузного, захватывает глубокие слои роговицы и более интенсивно в лимбальной зоне.
Строма роговицы всегда утолщена. Может наблюдаться застойный сосок зрительного нерва, а затем и его атрофия. Сетчатка и особенно парапапиллярная зона отечны, сосуды глазного дна расширены, стенки их утолщены. Переднезадняя ось глазного яблока увеличена у всех больных по сравнению с нормой на 2 -2,5 мм. Ни у одного больного не было выявлено повышения внутриглазного давления.
Синдром Гунтера выявляется преимущественно у мальчиков в возрасте 3 лет и старше. Ранние изменения скелета в виде увеличения размера черепа расценивают как признаки рахита. Тугоподвижность и изменения формы суставов становятся заметными к 3 годам, а иногда в более старшем возрасте. Изменения скелета в виде деформации грудной клетки и позвоночника выражены умеренно.
Изменения органа зрения при синдроме Гунтера характеризуются ранним увеличением роговицы и глазного яблока, но менее выраженным, чем при синдроме Гурлер. Помутнение роговицы выявляется к 4 - 6 годам, а иногда и позже. При биомикроскопическом исследовании выявляют, что наиболее интенсивное помутнение отмечается на 3 и 9 часах у лимба в глубоких слоях роговицы, строма которой утолщена.
В строме радужки, преимущественно на 3 и 9 часах, определяются новообразованные кровеносные сосуды. На глазном дне при офтальмоскопии обнаруживают расширенные кровеносные сосуды с утолщенной стенкой, а также застойные соски или частичную атрофию дисков зрительных нервов. Глазные симптомы предшествуют изменениям скелета и внутренних органов.
Синдром Санфилиппо характеризуется в первую очередь снижением интеллекта. Отмечается слабовыраженная гепатоспленомегалия. Отставания в росте не замечено. Имеются тугоподвижность суставов и изменение их формы.
Из глазных симптомов при синдроме Санфилиппо наблюдаются: гипертелоризм и небольшой латеральный проптоз. Роговица прозрачна, размер ее в норме. На глазном дне выявляют некоторое расширение калибра вен, т. е. картина синдрома отличается от таковой при мукополисахаридозах I и II типов.
Синдром Моркио отличается значительными деформациями скелета.
При синдроме Моркио происходят изменения в переднем отрезке глаз в ранних стадиях биомикроскоппчески характеризующихся дистрофией роговицы. Может отмечаться застойный сосок зрительного нерва.
Синдром Шейе внешне имеет много общего с инфекционным неспецифическим полиартритом. Основными характерными симптомами заболевания являются изменения формы мелких и средних суставов, их болезненность и тугоподвижность.
Изменения глаз при синдроме Шейе характеризуются признаками врожденной глаукомы, возникающими уже в ранних стадиях заболевания. Позднее, когда присоединяются боли и происходит деформация мелких суставов, появляются такие же изменения глаз, как при коллагенозном увейте, с выраженной дистрофией роговицы, которая сопровождается буллезным перерождением. В начальной стадии помутнение локализуется в зоне передней пограничной пластинки, причем более интенсивное оно у лимба, а затем распространяется на все слои роговицы.
Наличие у больных тотального помутнения увеличенной роговицы с утолщенной стромой на фоне выраженного отека конъюнктивы свидетельствует о мукополисахаридозе. Диагноз подтверждают результаты определения содержания КГАГ в моче больных. Необходимо отметить, что наряду с резко выраженными клиническими формами заболевания встречаются стертые формы мукополисахаридозов, при которых ведущими признаками являются только поражения глаз.
В заключение необходимо указать, что результаты офтальмологических исследований в сочетании с общими симптомами заболевания позволяют диагностировать мукополисахаридоз и уточнить его тип. При определении экскреции КГАГ с мочой у больных мукополисахаридозом выявляют их увеличение в десятки раз по сравнению с нормой.
В то же время суточная экскреция оксипролина у всех этих больных оказывается сниженной. Особенно высокое содержание КГАГ в моче наблюдается у больных с синдромом Гурлер. Биохимическое изучение фракционного состава КГАГ показывает, что наряду с различиями имеется много общего в качественном составе КГАГ при всех типах мукополисахаридозов. У больных с синдромом Гурлер и Гунтера увеличено также выделение гепарансульфата. Особенно высокая гепарансульфатурия наблюдается при синдроме Санфилиппо.
Кроме описанных синдромов, характеризующихся поражением соединительной ткани, в офтальмологической, педиатрической и неврологической практике особое место занимают синдромы Марфана, Маркезани, гомоцистинурии и др.
Синдром Марфана (долихостеномиелия) - врожденное заболевание, характерными признаками которого являются высокий рост, необычная длина конечностей, тонкие длинные паукообразные пальцы, слабое развитие мышечной ткани. В последующие годы заболевание было названо арахнодактилией.
Тип наследования синдрома Марфана аутосомно-доминантный. Характерным симптомом заболевания является эктопия хрусталиков. Описаны пороки развития и других органов при синдроме Марфана, однако большинство исследователей считают наиболее типичными признаками заболевания поражение глаз, деформацию скелета и нарушения деятельности сердечно-сосудистой системы.
Из поражений глаз, кроме эктопии хрусталика, наблюдаются мегалокорнеа, кератоконус, голубые склеры, высокая близорукость, отслойка сетчатки. Нередко при синдроме Марфана развивается глаукома.
Для объективизации диагноза синдрома Марфана применяют исследование КГАГ. При синдроме Марфана экскреция КГАГ с мочой увеличена в 2—3 раза по сравнению с нормой.
Наиболее ранним глазным проявлением синдрома Марфана можно считать гипоплазию стромы радужки, особенно ее пигментной зрачковой каймы.
Вероятно, в связи с недостаточным развитием дилататора у этих больных не всегда удается вызвать хороший медикаментозный мидриаз. Часто в ранних стадиях заболевания обнаруживают отростки радужки и остатки зрачковой мембраны. Эктопия хрусталика кверху, книзу, кнутри и кнаружи различной степени выраженности наблюдается у всех больных.
При офтальмоскопии у детей с пороком сердца на глазном дне обнаруживают расширенные и извитые вены. В более старшем возрасте выявляются близорукость разной величины, деструкция стекловидного тела по типу зернистой и фибриллярной, растяжение, деструкция и разрывы волокон ресничного пояска связок, более выраженные в нижних отделах экватора хрусталика. Изменения глазного дна проявляются также в виде дистрофии сетчатки и ее разрывов.

Из других глазных симптомов встречаются эпикантус, эмбриотоксон, кератоконус, косоглазие, катаракта, микро- и сферофакия, врожденная глаукома (табл. 32). Изменения на глазном дне отмечаются у детей старшего возраста.
Миопическая рефракция, о наличии которой свидетельствуют результаты эхобиометрии толщины хрусталика (на 1—2 мм больше по сравнению с нормой), обусловлена более высокой преломляющей способностью хрусталика вследствие изменения его сферичности под влиянием разрыва волокон ресничного пояска. Сагиттальная ось глаза нормальная. Не исключено развитие и усиление близорукости вследствие образования кератоконуса. Иначе говоря, имеются основания утверждать, что близорукость при синдроме Марфана имеет чисто рефракционную, но не осевую природу.
Результаты биохимических исследований свидетельствуют о том, что изменения органа зрения при синдроме Марфана связаны в основном с увеличением содержания коллагена, о чем свидетельствует оксинупролинурия. Увеличение экскреции КГАГ, особенно гиалуроновой кислоты, отягощает течение основного заболевания.
В клинико-офтальмологических исследованиях установлено, что очень сходная картина глазных изменений обнаруживается и при других заболеваниях, в частности при гомоцистинурии (табл. 33).

Эктопия в сочетании с другими симптомами были выявлены у больных с синдромом Маркезани. Эти больные маленького роста, череп у них имеет брахицефалическую форму, туловище, шея и конечности короткие. Мышцы и подкожная жировая клетчатка хорошо выражены. Тип наследования доминантный и аутосомно-рецессивный.
Поражения глаз при синдроме Маркезани характеризуются тем, что передняя камера в центре мелкая, а на периферии глубокая, а также дрожанием радужки по периферии. Отдельные участки радужки атрофичны.
Редко наблюдается аниридия. Хрусталик имеет шаровидную форму, уменьшен в размере. Хорошо видны волокна ресничного пояска. В некоторых случаях хрусталик смещен книзу. Наблюдаются случаи вывиха хрусталика в переднюю камеру или стекловидное тело.
При вывихе хрусталика в переднюю камеру возникает вторичная глаукома. В подобных случаях отмечается высокая близорукость (хрусталиковая). Изредка наблюдаются дегенерация и отслойка сетчатки, а также атрофия зрительного нерва.
Глазные изменения в виде истончения и повышенной прозрачности склер наблюдаются при синдроме В а н-д е р-Х е в е. Для заболевания характерна тугоухость. Синдром голубых склер относится к конституциональным особенностям соединительной ткани, обусловленным множественными генными дефектами; наследуется по аутосомно-доминантному типу.
Заболевание связано с изменением эластических и коллагеновых волокон. У 60% больных склеры имеют голубую окраску. Причиной изменения окраски склер является тонкая наружная оболочка глаза, через которую просвечивается сосудистая оболочка.
Серовато-синяя окраска склер наблюдается уже у новорожденных. Наблюдающаяся в норме у новорожденных голубоватая окраска склер к 6 мес постепенно, исчезает, и у здоровых детей они становятся белесоватыми.
Голубые склеры сочетаются с кератоконусом, кератоглобусом, мегалокорнеа, помутнениями роговицы по типу пятнистой дегенерации. Роговица истончена. Иногда часть склеры, непосредственно прилегающая к роговице, белая и называется «кольцо Сатурна». Редко наблюдается сочетание голубых склер с эктопией хрусталика.
Рефракцая чаще гиперметропическая.
Необходимо отметить, что глазные симптомы постоянно сопутствуют псевдомукополисахаридозу. При этом в возрасте до 3 лет развивается аннулярное помутнение роговицы, сопровождающееся васкуляризацией. Острота зрения постепенно снижается.
Необходимо отметить, что глазные симптомы постоянно сопутствуют псевдомукополисахаридозу. При этом в возрасте до 3 лет развивается аннулярное помутнение роговицы, сопровождающееся васкуляризацией. Острота зрения постепенно снижается.
У больных наблюдаются утолщение мелких суставов конечностей и позвоночника, их деформация и ограничение подвижности. Выражен остеопороз. Отмечаются задержка закрытия родничка и развития зубов, черты лица, характерные для больного гаргоилизмом, большой язык, кифосколиоз, изменения ЭЭГ.
Умственное развитие детей не страдает. Клиническая картина сходна с проявлениями мукополисахаридоза. Однако содержание КГАГ в моче у таких больных в норме. При гистохимическом исследовании в коже выявляются высокое содержание уроновой кислоты и метахромазия культуры фибробластов.
Прогноз в отношении зрения малоблагоприятный, но отмечен эффект от применения больших доз ретинола (витамина А).
Таким образом, глазные поражения при болезнях соединительной ткани характеризуются полиморфизмом.
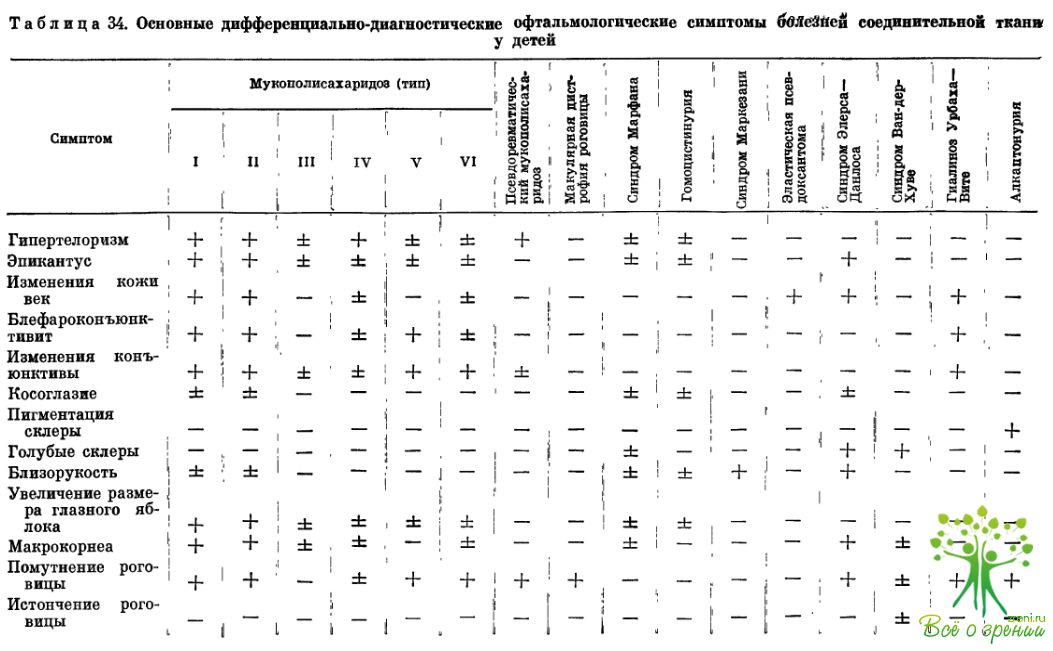

Изучение ранних офтальмологических симптомов позволяет дифференцировать отдельные формы заболеваний (табл. 34). Офтальмологические исследования помогают понять важные стороны патогенеза изучаемых заболеваний, уточнить роль изменений коллагена и основного вещества соединительной ткани в их развитии.
Изучение обмена КГАГ и оксипролина позволяет уточнить механизмы развития офтальмологических симптомов при болезнях соединительной ткани, а следовательно, принять соответствующие меры для предупреждения их возникновения и лечения.

Комментариев 0