Нейрометаболические заболевания | Детская офтальмология


Содержание:
Описание
Нейрометаболические заболевания или нейролипидозы относят к группе наследственных расстройств обмена веществ, в сочетании с поражением нервной системы. Эти расстройства характеризуются сопутствующей патологией органа зрения. Вариабельность симптоматики глазных заболеваний при этих нарушениях обусловлена вовлечением в процесс нервных элементов пигментного эпителия сетчатки и сосудистой оболочки, обладающих высоким уровнем обменных процессов.↑ Ганглиозидозы
GM 1 Ганглиозидозы
Инфантильный GM 1, тип I
- Дефицит р-галактозидазы.
- Вовлечение в процесс внутренних органов: увеличение печени, селезенки и почек.
- Грубые черты лица, фенотип Гурлер (Hurler).
- Появление на глазном дне симптома «вишневой косточки».
- Помутнение роговицы.
- Церебральная дегенерация, периодические припадки.
- Регрессия и смерть в раннем детском возрасте.
- Аутосомно-рецессивный тип наследования.
Поздний инфантильный GM 1, тип II
- Умственная и физическая деградация.
- Неизмененные черты лица.
- Припадки.
- Кифосколиоз.
- Аутосомно-рецессивный тип наследования.
Болезнь Тея-Сакса (Tay-Sachs), Тея-Сакса (GM2 тип I), Сандхофф (Sandhoffs) (GM2 тип II)
- Болезнь Тея-Сакса: дефицит гексозаминидазы А.
- Болезнь Сандхоффа: дефицит гексозаминидазы А и В.
- Деменция в первые, годы жизни.
- Извращенная реакция испуга на свет и звук.
- Увеличение размеров головы.
- Появление на глазном дне симптома «вишневой косточки», развитие атрофии зрительного нерва.
- Гибель ребенка в возрасте 3-4 лет.
- Аутосомно-рецессивный тип наследования.
↑ Болезнь Баттена (Batten) (нейронный цероидный липофузиноз)
Различают четыре основных формы болезни Баттена. При всех типах расстройства наблюдаются дегенарация сетчатки, регрессия и припадки. Офтальмологи чаще встречаются с ювенильной формой заболевания.
Ювенильная болезнь Баттена
Известна также как болезнь Баттена-Мейю (Batten-Mayou), болезнь Фогта-Шпильмейера (Vogt-Spielmeyer), болезнь Шпильмейера-Сьегрена (Spielmeyer-Sj?gren) и ювенильный нейронный цероидный липофузиноз.
- Прогрессирующее снижение зрения, появляющееся между 4 и 10 годами.
- Макулопатия «бычий глаз» (рис. 20.1).
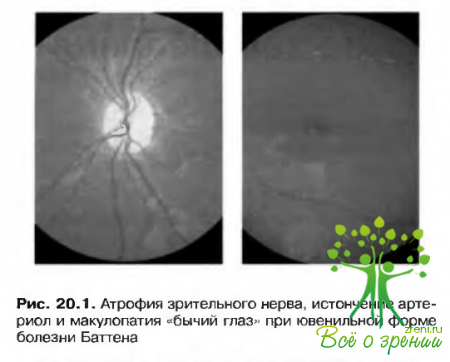
- Эксцентричный взгляд — больной стремится удержать взор вверх, используя таким образом нижнее поле зрения.
- В отдаленные сроки развивается тотальная дегенерация сетчатки.
- Патологические изменения электроретинограммы (ЭРГ), первоначально снижается волна В.
- К 3-летнему возрасту обычно наступает слепота.
- Умственная деградация, изменение поведения ребенка в период снижения зрения.
- Постепенная потеря навыков, регрессия и смерть на 3 десятилетии жизни.
- Припадки на 1-2 десятилетии жизни.
- Возможность предотвращения припадков.
- Аутосомно-рецессивный тип наследования.
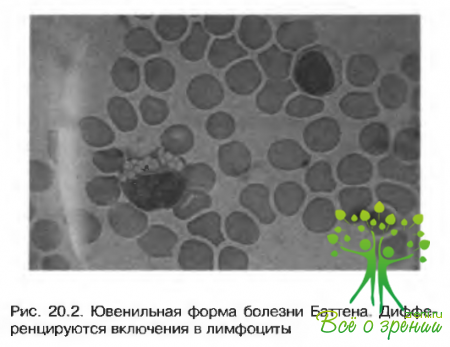
Диагностика
- Вакуолизация лимфоцитов в мазках периферической крови (рис. 20.2).
- Симптомы «пальцевых вдавлений» при исследовании электронной микроскопии ткани кожи и конъюнктнвы или лимфоцитов.
↑ Болезнь Ниманна-Пика (Метапп-Рюк)
[banner_centerrs] {banner_centerrs} [/banner_centerrs]
1 Группа
Встречаются детская, юношеская и взрослая формы заболевания. Классическая инфантильная форма проявляется в грудном возрасте. Заболевание обусловлено недостаточностью сфингомиелиназы.
Тип А: нейровисцеральная, инфантильная форма
- Возникает в первые 6 месяцев жизни.
- Гепатоспленомегалия.
- Регрессия.
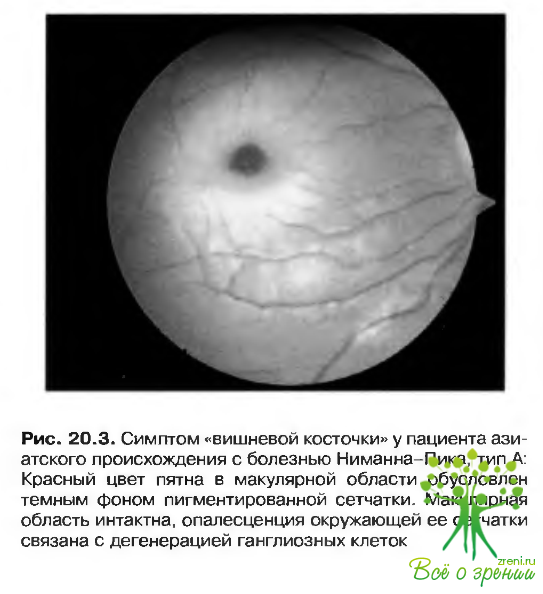
- Симптом «вишневой косточки» (рис. 20.3).?
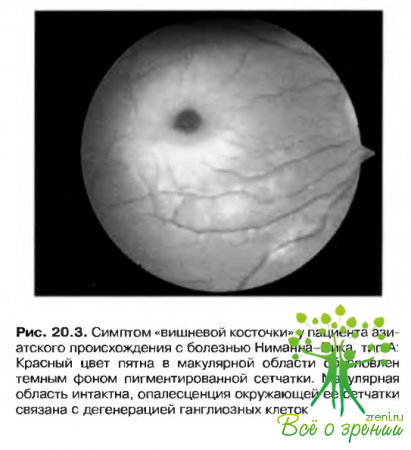
- Атрофия зрительного нерва возникает в отдаленные сроки.
- Аутосомно-рецессивный тип наследования.
Тип В: висцеральная форма
- Характерно позднее начало.
- Симптом «вишневой косточки» выражен слабо.
- Распространенная висцеромегалия.
- Инфильтративные изменения легких и костного мозга.
2 Группа
Для этой группы больных характерно нормальное содержание сфингомпелиназы в тканях.
Тип С: сроки начала заболевания вариабельны.
- У детей грудного возраста проявляется дисфункцией печени, желтухой, психомоторной задержкой развития и ранней смертью.
- У детей 4-6 летнего возраста заболевание проявляется умственной отсталостью, вертикальным супранукларным парезом, атаксией.
- Возможна манифестация в подростковом возрасте.
Наиболее распространенным патологическим изменением органа зрения при возникновении заболевания на первом десятилетии жизни является вертикальный супрануклеарный парез (рис. 20.4), в частности утрата вертикальных саккалических движений, особенно вниз. Характерен симптом «вишневой косточки».
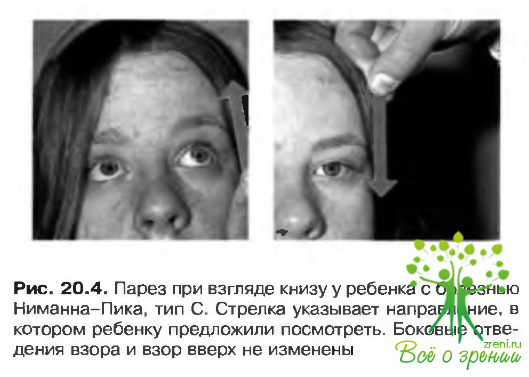
Диагноз ставят на основании наличия «пенистых» макрофагов в костном мозге и пограничных полиморфных мембран цитопластических образовании в нейронах. Тип наследования аутосомно-рецессивный.
Tип D
- Встречается редко.
- Аналогичен типу С.
- Обнаружен в Новой Шотландии.
↑ Болезнь Гоше (Gaucher)
- Дефицит глюкоцерсброзндазы.
- Наличие клеток Гоше в различных тканях.
- Аутосомно-рецессивный тип наследования.
Тип I
- Гепатоспленомегалия.
- Пингвекулы.
- Дегенграния центральной нервной системы возникает редко.
- Гало в макулярной области возникает редкоко.
- Позднее начало.
Тип II
- Манифестация с рождения
- Припадки.
- Симптом «вишневой косточки».
- Задержка развития, регрессия и смерть в раннем лете ком возрасте.
Тип III
- Возникает в период с рождения до 14 лет.
- Снижение интеллекта, припадки.
- Вертикальный сунрануклеарный парез.
- Косоглазие.
↑ Дефицит нейраминидазы
Сиалидоз тип I: симптом «вишневой косточки» — миоклонический синдром.
- Симптом «вишневой косточки», постепенное снижение зрения.
- Прогрессирующая миоклония.
Сиалидоз тип II
- Фенотип Гурлер.
- Регрессия.
- Помутнение роговицы, симптом «вишневой косточки».
Памятка 20.1.
Глазодвигательные расстройства при нейрометаболических заболеваниях
- Парез вертикального взора («апраксия»):
- а. болезнь Ниманна-Пика, тип С;
- б. болезнь Гоше.
- а. болезнь Ниманна-Пика, тип С;
- Нистагм:
- а. сиалидоз, тип I;
- б. болезнь Кэнэвана (Canavan);
- в. болезнь Пелицеуса-Мерцбахера (Pelizaeus- Merzbacher);
- г. все заболевания, ведущие к слепоте.
- а. сиалидоз, тип I;
- Хаотические движения глаз:
- а. органическая ацидемия;
- б. расстройства цикла образования мочевины;
- в. болезнь Ли (Leigh).
- а. органическая ацидемия;
- Косоглазие: в большинстве случаев наблюдается снижение зрения.
↑ Мукополисахаридоз
Раньше детские офтальмологи редко сталкивались с этим состоянием (табл. 20.1). Увеличение продолжительности жизни больных благодаря внедрению пересадки костного мозга явилось причиной проявления симптоматики со стороны органа зрения при мукополисахарндозе.

Разные формы заболевания имеют общие черты, присутствующие в различных сочетаниях.
Помутнение роговицы при мукополисахаридозе
Роговица утолщена, ее клетки, строма и межклеточное пространство диффузно инфильтрированы гликозаминогликанами. Помутнение роговицы носит гомогенный характер (рис. 20.5).
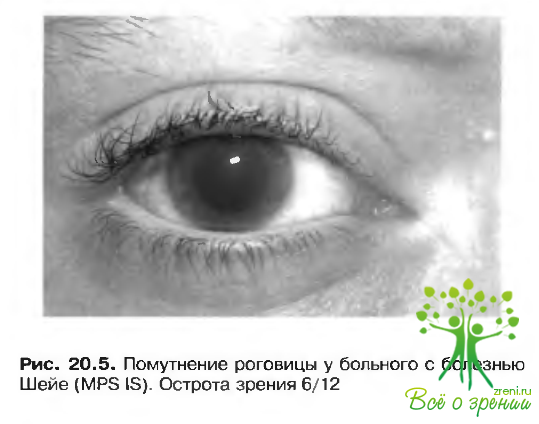
Возможно полное или частичное рассасывание помутнения после пересадки костного мозга. При отсутствии эффекта показана пересадка роговицы, но необходимо учитывать, что состояние роговицы не является основной причиной снижения зрения. Помутнение роговицы не характерно для болезни Гунтера и протекает сглажено при синдроме Санфилиппо.
Дегенерация сетчатки при мукополисахаридозе
Присутствует при всех формах заболевания, кроме болезни Моркио. Как правило, диагностируется только по данным ЭРГ, так как офтальмоскопия глазного дна из-за помутнения роговицы обычно недоступна. Симптоматика вялая, часто начинается с затруднения ночного зрения.
Нейропатия зрительного нерва при мукополисахаридозе
Нейропатия зрительного нерва обусловлена его прямой инфильтрацией гликозаминогликанами. Существуют дополнительные причины возникновения двустороннего застойного соска, как при повышенном, так и при нормальном внутричерепном давлении. Одна из этих причин — компрессия основания зрительного нерва утолщенной склеральной оболочкой. Нейропатия зрительного нерва редко наблюдается при синдроме Санфилиппо и болезни Моркпо. Атрофия зрительного нерва может быть следствием глаукомы.
Глаукома
Глаукома частый спутник мукополасахаридоза и ее выявление является важной задачей. Возникает в связи с блокадой путей оттока гликозаминогликанами. Помутнение роговицы осложняет диагностику и наблюдение за динамиком процесса.
Роль офтальмолога в лечении больных мукополисахаридозами
- Удостоверьтесь, что острота зрения пациента не может быть повышена за счет подбора очков или специальных приспособлений.
- Пересадка роговицы целесообразна при отсутствии дегенерации сетчатки, глаукомы и нейропатии зрительного нерва.
- Наблюдение за пациентом для исключения глаукомы и атрофии зрительного нерва.
- Диагностика отека диска зрительного нерва, при необходимости, коррекция повышенного внутричерепного давления медикаментозными средствами, шунтированием или фепестрацией оболочек зрительного нерва.
- Наблюдение за состоянием роговицы, сетчатки и других внутриглазных структур после пересадки костного мозга.
- Нейрофизиологическое исследование помогает диагностике патологии сетчатки и зрительного нерва, а также обеспечивает возможность динамического наблюдения.
↑ Муколипидозы
Редко встречающиеся расстройства, имеющие некоторые общие признаки с мукополисахаридозами и нейролинидозами. С точки зрения детской офтальмологии имеет значение только муколипидоз IV.
Муколипидоз IV
- Помутнение или мутовчатость роговицы, возникающие на 2-м году жизни.
- Помутнение роговицы захватывает в основном эпителиальный слой.
- Существуют формы заболевания с мягким течением.
- Дегенерация сетчатки, катаракты.
- Регресс развития, гипотония и, через несколько лет, смерть.
- Аутосомно-рецессивный тип наследования.
Маннозидоз
- Нежное помутнение роговицы.
- Спицевидные помутнения в задних кортикальных слоях хрусталика.
- Грубые черты лица, изменения костей, регрессия.
- Аутосомно-рецессивный тип наследования.
Фукозидоз
- Умственная и физическая деградация.
- Грубые черты лица.
- Дисплазия костей.
- Аутосомно-рецессивный тип наследования.
- Извитость сосудов конъюнктивы и сетчатки.
- Макулопатия «бычий глаз».
- Помутнения роговицы.
Болезнь Фабри (Fabry)
- Диффузная ангиокератома туловища
- Выраженная периферическая ненропатия.
- Заболевания почек.
- Извитость сосудов конъюнктивы.
- Отек век.
- Мутовчатость роговицы (рис. 20.6).

- Помутнения хрусталика.
- Нистагм.
- Атрофия зрительного нерва.
- Х-сцепленный рецессивный тип наследования.

↑ Лейкодистрофии
Метахроматическая лейкодистрофия
- Прогрессирующие двигательные расстройства и изменение поведения.
- Периферическая нейропатия.
- Первичная атрофия зрительного нерва в каждом 3-м случае.
- Симптом «вишневой косточки» и последующая атрофия зрительного нерва.
- Аутосомно-рецессивный тип наследования.
- Диагностика:
- а. наличие внутриклеточных мочевых метахроматических включений;
- б. дефицит арилсульфатазы.
- а. наличие внутриклеточных мочевых метахроматических включений;
Болезнь Краббе (Krabbe)
- Дефицит галактоцереброзндазы.
- Регрессия с раннего детства.
- Раздражительность, утрата навыков.
- Повышенная реакция на любой раздражитель.
- Периферическая нейропатия.
- Атрофия зрительного нерпа.
- Центральная слепота.
Адренолейкодистрофия
- Центральная слепота в сочетании с появлением пернвентрикулярных зон низкой плотности в задней области белого вещества коры головного мозга (рис. 20.7) на сканограммах компьютерной томографии (KT) или магнитно-резонансной томографии.
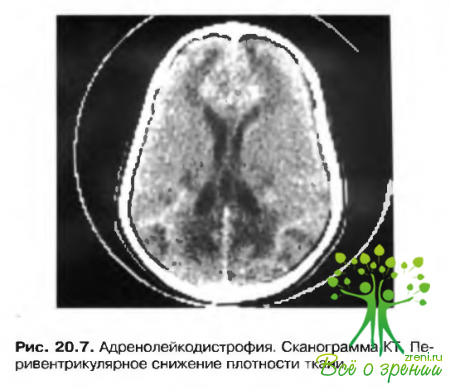
- Болезнь Аддисона (Addison) с гиперпигментацией.
- Атрофия зрительного нерва.
- Х-сцепленный рецессивный тип наследования.
- Регрессия и смерть в течение нескольких лет от начала заболевания.

Органические ацидемии
Например, болезнь «кленового сиропа», пропионовая ацидемия, глютаровая ацидурия и т.д.
- Метаболические ацидозы.
- Быстрая деградация.
- Кетоз.
- Задержка развития.
- Припадки.
- Парез взора.
- Нистагм.
- Птоз.
Болезнь Менке (Menke's)
- Х-сцепленный рецессивный тип наследования.
- Задержка развития с рождения.
- Патология волос (болезнь курчавых волос).
- Редкие волосы на бровях.
- Густые ресницы.
- Дистрофия сетчатки с патологическими изменениями ЭРГ.
- Извитость сосудов сетчатки.
- Демиелинизация зрительного нерва.
Фенилкетонурия
- Грубые нарушения рефракции.
- Катаракта.
- Постепенная умственная деградация.
- Бледная шероховатая кожа.
- Белокурые волосы и голубые глаза.
- Аутосомно-рецессивный тип наследования.
Памятка 20.2.
Симптом «вишневой косточки»
- GM2 типы I, II и III.
- GM1 тип I.
- Болезнь Ниманна-Пика:
- а. группа 1, типы А и Б;
- б. группа 2, тип С (иногда при болезни Гоше, тип II, реже тип I).
- а. группа 1, типы А и Б;
- Метахроматическая лейкодистрофия.
- Болезнь Фарбера (Farber).
- Муколипидоз тип III.Сиалидоз типы I и II.

Комментариев 0