Стекловидное тело | Детская офтальмология


Содержание:
Описание
↑ Развитие
Первичное стекловидное тело возникает приблизительно на 6-й неделе внутриутробного развития и состоит из мезодермальных клеток, коллагеновых волокон, гиалоидных сосудов и макрофагов. Вторичное стекловидное тело формируется на втором месяце и содержит компактную фибриллярную сеть, гиалоциты, моноциты и некоторое количество гиалуроновой кислоты. В конце третьего месяца внутриутробного развития формируется третичное стекловидное тело, представляющее собой сгущенное скопление коллагеновых волокон между экватором хрусталика и диском зрительного нерва. Это предшественник основного стекловидного тела и зонулярных связок хрусталика. Ближе к концу четвертого месяца внутриутробного развития первичное стекловидное тело и гиалоидная сосудистая сеть атрофируются, превращаясь в прозрачную узкую центральную зону, относящуюся к клокетовому каналу. Персистенция первичного стекловидного тела — важный фактор при некоторых пороках развития стекловидного тела.
↑ Пороки развития
Персистирующая гиалоидная артерия
Персистенция гиалоидной артерии наблюдается более чем у 3% здоровых доношенных младенцев. Она практически всегда обнаруживается на 30-й неделе внутриутробного развития и у недоношенных детей при скрининге с целью выявления ретинопатии недоношенных. Остатки задней части этой гиалоидной сети могут проявляться как приподнятая масса гиалоидной ткани на диске зрительного нерва и называются соском Бергмейстера. Передние остатки, выглядят прилипшими к задней капсуле хрусталика, и называются пятном Миттендорфа.
Персистирующая гиперплазия первичного стекловидного тела (ПГПС)
ПГПС — врожденная патология глаза, связанная с нарушением регресса первичного стекловидного тела. Встречается редко и имеет односторонний характер. В большинстве сообщений о двусторонней и семейной ПГПС речь по-видимому идет о различных синдромах, включающих витреоретинальную дисплазию.
Классические признаки ПГПС:
- фиброзная мембрана, сращенная с задней поверхностью хрусталика;
- микрофтальм;
- мелкая передняя камера;
- расширение сосудов радужки;
- васкуляризированная ретролентальная мембрана, вызывающая тракцию цилиарных отростков (рис. 16.1).

Другие глазные симптомы, сопутствующие ПГПС, встречаются редко и включают:
- мегалокорнеа;
- аномалию Ригера;
- аномалию диска «утреннее сияние».
Хотя была описана так называемая задняя форма ПГПС, остается неясным, чем отличаются эти случаи от серповидных складок или витреоретинальной дисплазии.
Лечение ПГПС обычно направлено на профилактику глаукомы и фтизиса глазного яблока. Удаление хрусталика и задней мембраны может предупредить развитие глаукомы даже на глазу с низким функциональным прогнозом. Однако необходимо заметить, что есть сообщения о возникновении глаукомы после ленсвитрэктомии. Некоторые авторы отмечают возможность значимого повышения остроты зрения при ранней хирургии и активном плеоптическом лечении.
Кисты стекловидного тела
Кисты стекловидного тела могут быть как врожденными, так и приобретенными (рис. 16.2). Происхождение кист неизвестно, но так как внутри кисты иногда присутствуют кровеносные сосуды, предполагают, что кисты берут начало от остатков гиалоидной артерии.
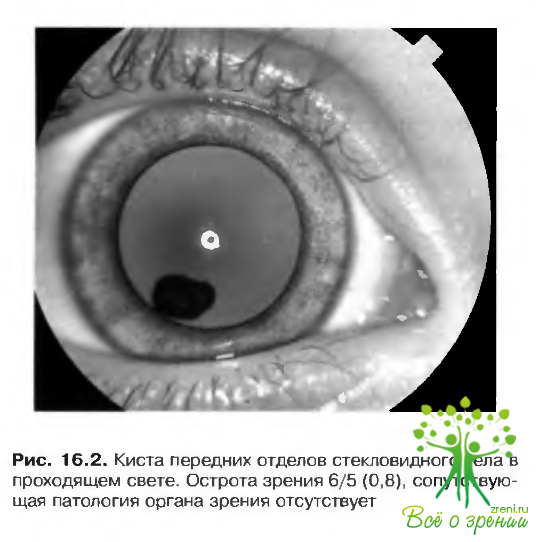
Приобретенные кисты могут сочетаться с:
- токсоплазмозом;
- токсокарозом;
- ювенильным ретиношизисом.
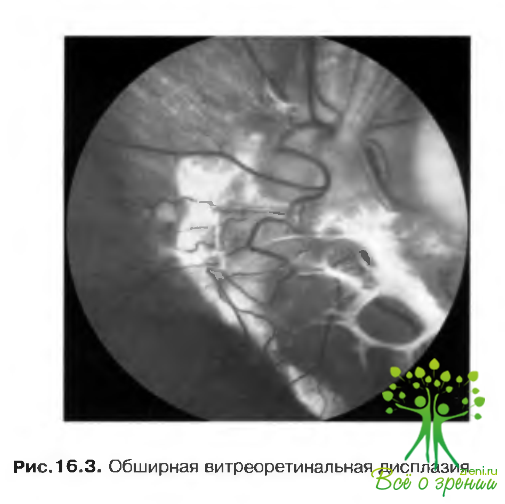
Витреоретинальная дисплазия
Витреоретинальная дисплазия проявляется как в виде изолированной патологии, так и в сочетании с разнообразными системными нарушениями (рис. 16.3).
Трисомия 13 (синдром Патау [Patau])
Выраженная двусторонняя глазная патология, наблюдающаяся практически во всех случаях трисомии 13 и включающая следующие симптомы:
- обширную сетчатки;
- фиброз стекловидного тела;
- микрофтальм;
- колобомы;
- циклонию:
[banner_centerrs] {banner_centerrs} [/banner_centerrs] - катаракты;
- помутнения роговицы;
- глаукому.
Характерна также тяжелая системная патология, обусловливающая малый срок жизни.
Болезнь Норри (Norrie) (болезнь Андерсена-Вабурга [Andersen-Warburg])
Болезнь Норри — сцепленное с Х-хромосой рецессивное заболевание, при котором могут присутствовать потеря зрения, слуха и умственная отсталость. Описаны три случая значительного снижения зрения у женщин при болезни Норри. Определен отвечающий за болезнь Норригеи, кодирующий аминокислотами. В семьях-носителях пораженного гена обнаружены дополнение радикала этого гена.
Идентификация гена болезни Норри позволит диагностировать носителя и пораженных лиц.
Глазные проявления включают:
- двусторонние складки сетчатки:
- отслойку сетчатки;
- витрсальные геморрагии;
- двусторонний ретролентальный конгломерат фиброзной ткани (витреоретинальная дисплазия);
- физис глазного яблока (рис. 16.4).
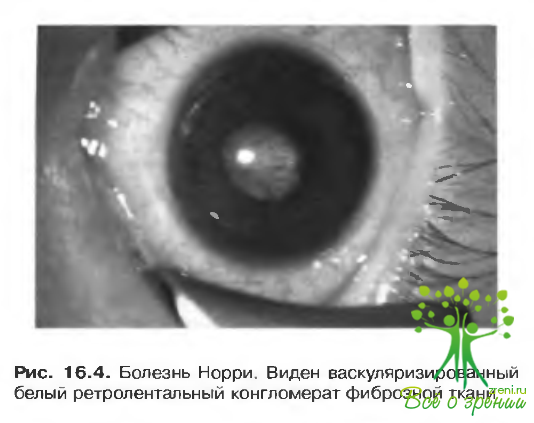
Практически все пораженные — мужчины, слепые от рождения или с раннего детства. Около 25°о пораженных лиц страдают умственной отсталостью, а в 33"о случаев развивается нейросенсорная тугоухость.
Инконтиненция пигмента (недержание пигмента) (Синдром Блоха-Сульцбергера)
Редкое заболевание, вовлекающее в процесс эктодермальные структуры (такие, как глаза, кожа, зубы и центральная нервная система). Пораженные лица с детства страдают везикулярной, которая эволюционирует в характерную патологическую пигментацию. С возрастом она становится менее заметной (рис. 16.5).

Наследуется как сцепленный с Х-хромосомой доминантный (с внутриутробной гибелью плодов мужского иола) синдром, соединенный на хромосомной карте с регионом Xq28. Передачи заболевания от мужчины к мужчине не зарегистрировано, хотя отмечены случаи передачи от мужчины к женщине.
Глазная патология включает
- патологическую васкуляризацию сетчатки извитость сосудов, облитерация капилляров, периферические или венозные шунты;
- преретинальный фиброз;
- тракционную отслойку сетчатки;
- ретинальные геморрагии;
- макулярный отек;
- катаракту;
- атрофию зрительного нерва;
- нистагм и косоглазие.
Можно избежать отслойки сетчатки у пораженных женщин, если лечение, заключающееся в криотерапии или фотокоагуляции, начато сразу после выявления сосудистой патологии сетчатки.
Синдром Уокера-Вабурга (Walker-Warburg)
Заболевание наследуется на аутосомно-рецессивному типу и включает:
- витреоретинальную дисплазию и атрофию;
- порок развития мозжечка;
- врожденную мышечную дистрофию;
- Депди-Уокера (Dandy-Walker) порок развития;
- лицефалоцеле (рис. 16.6).

Другие синдромы, включающие витреоретинальную дисплазию:
- остеопороз на фоне псевдоглиомы:
- а. умственная отсталость
- б. наследуется по аутосомно-рецессивному типу;
- а. умственная отсталость
- глазо-небно-церебральная задержка развития с сочетанной патологией:
- а. микроцефалия;
- б. умственная отсталость;
- в. расщепленное небо;
- г. маленький рост;
- д. сочетается с расстройствами, напоминающими ПГПС, как односторонними, так и двусторонними.
- а. микроцефалия;
↑ Наследственная витреоретинальная дегенерация и отслойка сетчатки
Синдром Стиклера (Stickler) (наследственная прогрессирующая артро-офтальмопатия)
Описан Стиклером как состояние, включающее высокую миопию и раннюю отслойку сетчатки в сочетании с быстро возникающими дегенеративными изменениями суставного хряща. Наследуется как аутосомно-доминантный признак и по некоторым сообщениям более распространен, чем синдром Марфана.
Глазные проявления включают:
- врожденную стационарную миопию высокой степени;
- разжижение стекловидного тела;
- мембраны и пленки в стекловидном теле;
- периваскулярные пигментные изменения сетчатки;
- хориоретинальную дегенерацию и разрывы сетчатки;
- первичную открытоугольную глаукому;
- предстарческую ядерную катаракту;
- периферическую кортикальную катаракту с помутнениями в виде спиц.
Системные нарушения включают:
- прогрессирующую артропатию;
- заячью губу и волчью пасть;
- патологически измененные черты лица;
- нейросенсорную тугоухость.
Клинически представляет собой разнородную группу доминантных заболеваний нескольких подтипов, включая марфаноподобный вариант, вариант с аномалией Пьера-Робина (Pierre-Robin) и больных с классическим фенотипом — плоским лицом и седловидным носом. Генетический код одной из форм синдрома Стиклера идентифицирован на хромосомной карте как 12ql4. Более редкие мутации идентифицированы как COL2AI. Эти больные нуждаются в тщательном обследовании и профилактической лазер- или криотерапии любого разрыва сетчатки. Лечение отслоек сетчатки комплексное, результаты его часто неудовлетворительные.
Синдром Книста (Kniest)
Этот синдром близок к синдрому Стиклера и также наследуется по аутосомно-доминантному типу.
Сопутствующая этому синдрому системная патология включает:
- дисплазию костей;
- выступающие утолщенные суставы;
- укороченное туловище;
- деформацию грудины;
- плоское лицо с седловидным носом;
- волчью пасть.
Глазные симптомы:
- миопия высокой степени;
- катаракта;
- глаукома;
- отслойка сетчатки.
Лечение подобно таковому при синдроме Стиклера.
Синдром Вагнера (Wagner)
Изолированный глазной синдром с отсутствием системных проявлений.
Характерные черты включают:
- миопию средней степени;
- деструкцию стекловидного тела;
- преретинальную бессосудистую мембрану;
- изменения сосудов сетчатки с образованием периваскулярных муфт и периваскулярной пигментацией;
- прогрессирующую атрофию сетчатки и сосудистой оболочки;
- прогрессирующую вплоть до затухания электроретинограммы (ЭРГ) дегенерацию сетчатки;
- отсутствие отслойки сетчатки;
- катаракты.
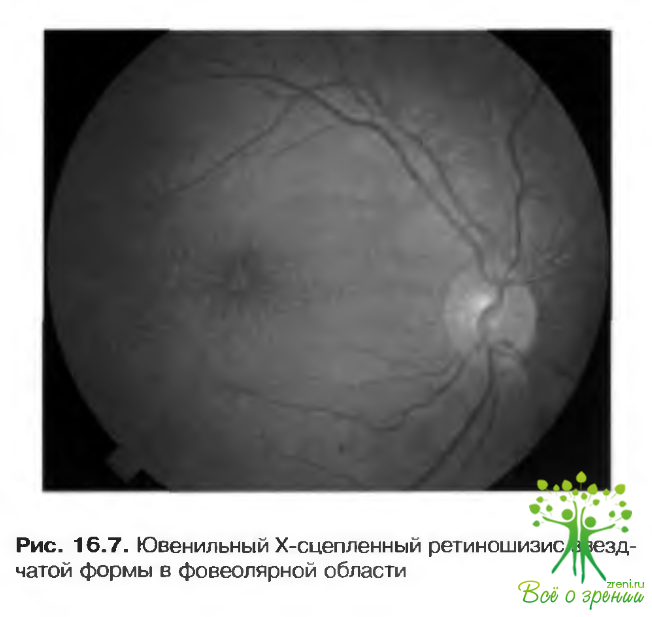
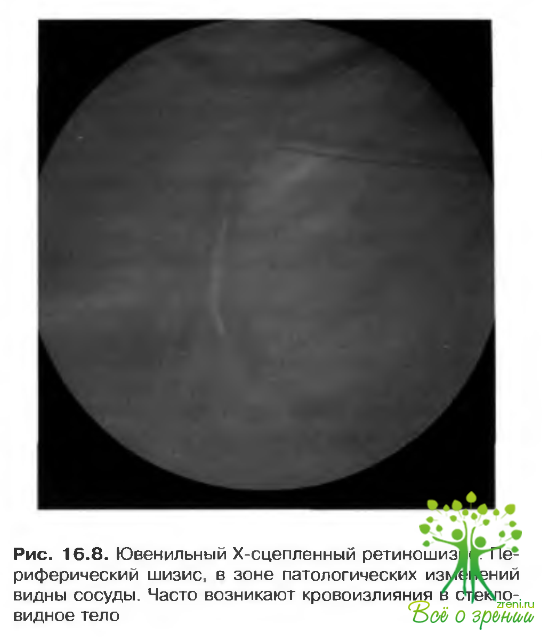
Ювенильный Х-сцепленный ретиношизис (врожденный наследственный ретиношизис)
Заболевание, не имеющее системных проявлении, наследуется как Х-сцепленный рецессивный признак. Патологический ген локализован в зоне Хр22. У женщин-носителей не выявляется патологических изменений.
Характерные особенности офтальмостатуса включают:
- Фовсолярный ретиношизис — наиболее постоянное явление, встречающееся в 50-ти случаев, может быть обнаружен даже в раннем детстве (рис. 16.7). Острота зрения, как правило, находится в пределах 6 12-6/36 (0.16-0,5).

- Ретиношизис в сегменте наблюдается у 40-50% больных. Разрыв появляется в слое нервных волокон. При повышении проницаемости сосудистой стенки может начаться кровотечение (рис. 16.8), при разрыве сосуда появляются интравитреальные геморрагии.

- Витрсальные мембраны (волокнистые уплотнения в стекловидном теле) обычно возникают над зонами периферического ретиношизиса.
- Задняя отслойка стекловидного тела.
- Пигментная ретинопатия, периваскулярные муфты, обтурация капилляров.
- Патологически измененная ЭРГ снижена, появляются нарушения волны В, при этом волна А остается неизмененной.
Заболевание относят к патологии Клеток Мюллера (Mueller), обусловливающей изменения как в макулярной области, так и на периферии сетчатки.
Болезнь Гольдманна-Фавра (Goldmann-Favre)
Редкое аутосомно-рецессивное заболевание без системных проявлений.
Особенности офтальмо- статуса включают:
- ретиношизис;
- прогрессирующую катаракту:
- пигментную хориоретинальную дегенерацию;
- разжижение стекловидного тела;
- кистозные изменения в макулярной области. Снижение зрения связано в основном с центральным ретиношизисом, возникающим в самом начале заболевания; острота зрения 6/36 (0,16) нередко встречается даже в детском возрасте.
Семейная экссудативная витреоретинопатия
Термин «семейная экссудативная витреоретинопатия» используют для описания группы на-следственных заболеваний, характеризующихся патологической васкуляризацией сетчатки в совокупности с экссудацией, неоваскуляризацией и тракционной отслойкой сетчатки. Клинические проявления могут имитировать ретинопатию недоношенных. И действительно, несколько сообщений о ретинопатии недоношенных у доношенных детей имели отношение именно к этому заболеванию.
Существует две основных формы.
- Аутосомно-доминантная форма — ген локализован на длинном плече хромосомы.
- Х-сцепленная рецессивная форма — ген локализован в одной из двух зон — пли в Хр21.3, или в ХрН. Может быть аллельной с болезнью Норри.
Аутосомно-доминантная экссудативная витреоретинопатия
Прогрессирующая витреоретинопатия — заболевание, для которого типично тупиковое окончание сосудов вдоль зубчатой линии па экваторе сетчатки п отсутствие системной патологии. Оно сопровождается высокой частотой клинических проявлений среди членов одной семьи.
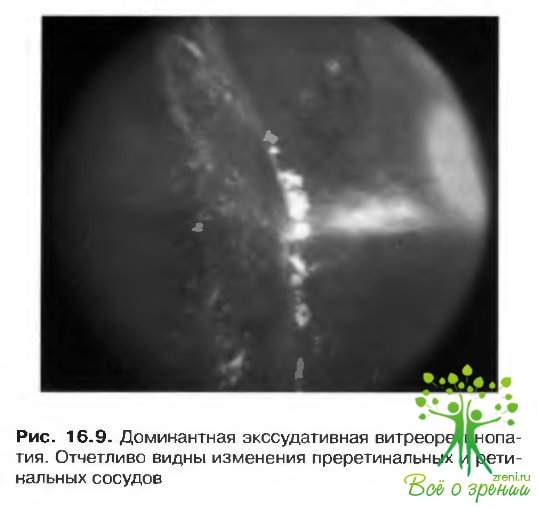
Офтальмостатус при этой патологии включает:
- периферические изменения сосудов сетчатки (рис. 16.9):
- а. расширение сосудов;
- б. извитость сосудов;
- с. артериовеиозные шунты;
- д. обтурация капилляров;
- е. периферическая пеоваскуляризация;
- а. расширение сосудов;
- пеоваскуляризацию диска зрительного нерва;
- эктопию макулы;
- тракционную отслойку сетчатки;
- макулярный отек:
- субретинальный экссудат.
Чаще всего отслойка сетчатки возникает в первые 10 лет жизни, в более старшем возрасте болезнь развивается очень медленно.
Х-сцепленная семейная экссудативная витреоретинопатия
Редкое расстройство, практически неотличимое от тяжелых форм доминантной экссудативном интесоретинопатии. Отличается очень ранним началом и плохим прогнозом. Заболевание с болезнью Норрн.
Аутосомно-доминантная витреоретинохориоидопатия
Это состояние отличается патологической стадией между варикозными венами и зубчатой линией по всей окружности. Задняя граница четко выражена возле экватора. У детей младшего школьного возраста возникают такие осложнения, как нпетозный макулярный отек и кровоизлияния в стекловидное тело. Для этого заболевания не характерно сочетание с системными расстройствами.
Наблюдается следующая офтальмологическая симптоматика:
- изменения сосудов сетчатки:
- а. сужение артерий;
- б. венозная непроходимость;
- в. повышение проницаемости стенок сосудов:
- а. сужение артерий;
- диффузное разжижение стекловидного тела;
- предстарческая катаракта;
- макулярный отек;
- кровоизлияния в стекловидное тело и отслойка сетчатки;
- сохранение нормальных параметров ЭРГ у большинства больных;• патологическое изменение Э01.
Аутосомно-доминантная неоваскулярная воспалительная витреоретинопатия
Редкое аутосомно-доминантное заболевание характеризуется обтурацией сосудов сетчатки и неоваскуляризацией, которые могут стать причиной кровоизлияний в стекловидное тело и тракционной отслойки сетчатки. Недавно был выделен и локализован в хромосоме ген, ответственный за возникновение процесса. В отличие от аутосомно-доминантной витреоретинохориоидопатии, заболевание склонно к прогрессированию; ЭРГ характеризуется с избирательным отсутствием волны В. Приблизительно в 20% случаев развивается отслойка сетчатки.
Эрозивная витреоретинопатия
Аутосомно-доминантное расстройство со следующими признаками:
- куриная слепота;
- прогрессирующее сужение полей зрения;
- патологические изменения в стекловидном теле;
- прогрессирующая пигментная атрофия сетчатки;
- отслойка сетчатки;
- распространенная дисфункция палочек и колбочек, регистрируемая на ЭРГ.
Функциональный прогноз плохой, у взрослых часто возникает отслойка сетчатки.
Аутосомно-доминантная инеевидная дегенерация (наследственная витреоретинальная инеевидная дегенерация Мгове)
Своим названием это заболевание обязано наличию белых, похожих на снежные хлопья помутнений, размером 100-200 мк. Помутнения появляются в зонах, напоминающих следы от давления, окруженные бледным ореолом. До 20-летнего возраста они обычно не выявляются.
Различают следующие, четко дифференцированные стадии:
- 1 стадия. Обширные области, напоминающие следы от давления, окруженные бледным ореолом.
- 2 стадия. Помутнения в виде снежинок, занимающие пространство от экватора до зубчатой линии сетчатки.
- 3 стадия. Образование периваскулярных муфт на сетчатке в зоне инеевидной дегенерации.
- 4 стадия. Прогрессирование пигментных изменений в зоне инеевидной дегенерации.
Памятка 16.1.
Кровоизлияния в стекловидное тело
Кровоизлияния в стекловидное тело возникают при различных заболеваниях, включая:
- ПГПС;
- Х-сцепленный ювенильный ретиношизис;
- семейную экссудативную витреоретинопатию, аутосомно-доминантную витреоретинохориоидопатию, аутосомно-доминантную воспалительную витреоретинопатию — вторично по отношению к неоваскуляризации;
- синдром Стиклера;
- гемангиобластому сетчатки;
- кавернозную гемангиому;
- болезнь Илза (Eale's);
- болезнь Коатца (Coats);
- геморрагический диатез.
В большинстве случаев при кровоизлияниях в стекловидное тело ограничиваются консервативным лечением, хотя в раннем детстве, из-за опасности развития депривационной амблиопии, может потребоваться витрэктомия.
По мере прогрессирования заболевания возникает сужение полей зрения, повышение палочкового порога при темновой адаптации, снижение амплитуды волны В скотопической ЭРГ. Сопутствующая системная патология отсутствует.
Приобретенные заболевания стекловидного тела
Воспалительные заболевания стекловидного тела
В детском возрасте встречаются следующие воспалительные заболевания стекловидного тела:
- ювенильный парепланит;
- срединный увеит;
- васкулит сетчатки;
- ювенильный ревматоидный артрит;
- саркоидоз;
- туберкулез;
- токсоплазмоз;
- токсокароз;
- острый некроз сетчатки.
Хотя у детей стекловидное тело редко поражается бактериальной инфекцией, выделяют три различных ситуации с наличием патогенной микрофлоры:
- Последствия глазных операций (включая хирургию косоглазия).
- Последствия проникающей травмы.
- Эндогенный метастатический эндофтальмит — возникает в сочетании с синдромом приобретенного иммунодефицита, менингитом и у пациентов с неблагополучием иммунной системы.
Диссеминация опухоли в стекловидном теле
Диссеминация опухоли в стекловидном теле характерна для ретинобластомы. Она может также возникать при лейкемии. Другие случаи диссеминации чрезвычайно редки.

Комментариев 0